abcsite.free.fr
![]()
Chimie Organique
|
|
8. Les composés carbonyles: le groupement C=O
| Dans ce chapitre | |
| 8.1. | Aldéhydes et cétones |
| Modèles Moléculaires en format .pdb: Aldéhydes | |
| Modèles Moléculaires en format .pdb: Cétones | |
| 8.2. | Énols et énones |
| 8.3. | Les acides carboxyliques |
| Modèles Moléculaires en format .pdb: Acides carboxyliques | |
| 8.4. | Diacides |
| 8.5. | Les dérivés des acides carboxyliques |
| Modèles Moléculaires en format .pdb: Amides | |
| Modèles Moléculaires en format .pdb: Esters | |
| 8.6. | Autres groupes fonctionnels contenant des liaisons multiples carbone-azote |
| Modèles Moléculaires en format .pdb: Composés Nitro | |
| 8.7. | Composés dicarbonylés et autres composés bifonctionnels |
| 8.8. | Exercices de nomenclature |
8.1 Aldéhydes et cétones (Vollhardt chapitre 17)
Les fonctions aldéhyde et cétone ont le même groupe fonctionnel >C=O, appelé le groupe carbonyle. Ils ne diffèrent que par le nombre de groupes alkyles ou aryles qui lentourent, ou encore par le degré de substitution du carbone fonctionnel.
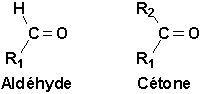
Les aldéhydes et cétones sont des composés carbonylés, et présentent beaucoup danalogies, mais la tradition a cependant maintenu cette distinction de deux fonctions distinctes.
On représente souvent les aldéhydes par RCHO. Il faut faire attention cependant de ne pas écrire RCOH, qui suggère un groupement alcool. De même, les cétones sont représentés par RCOR ou RCOR. Il faut garder à lesprit quil ne sagit pas dun enchaînement RCOR, qui est un éther.
Les aldéhydes et les cétones peuvent être considérés comme des dérivés oxydés des alcools.
Comme les alcools, loxygène carbonylique porte 2 doublets libres, ce qui explique le caractère légèrement basique des groupes carbonyles.
8.1.1. Nomenclature des aldéhydes et cétones (Vollhardt 17.1, Favre 5.1 et 5.2)
On nomme les aldéhydes et les cétones en changeant le e du nom de lhydrure parental par al et one respectivement. Lorsquen présence dun groupement plus important, ces deux groupes fonctionnels utilisent le préfixe oxo. Lorsque lon rencontre des polyaldéhydes, on utilise le suffixe carbaldéhyde. Lorsque le groupe CHO nest pas porté par la chaîne principale, on utilise le préfixe formyle.
Étant donné quun aldéhyde est toujours en bout de chaîne, il ne nécessite pas dindice de position, même lorsque lon parle de dialdéhyde. Il est à noter que le carbone dun groupement aldéhyde est le numéro 1, ce qui fixe automatiquement la numérotation de la chaîne.
Une cétone nécessitera toujours la présence dun indice de position. Enfin, notons quon dit un aldéhyde et une cétone.
8.1.2. Caractères Physiques
Le méthanal,
plus connu sous le nom de formaldéhyde, HCHO, est gazeux dans les conditions ordinaires, et lacétaldéhyde CH3CHO est un liquide extrêmement volatil, bouillant à 21°C. Mais tous les autres aldéhydes et cétones sont des liquides, ou des solides dans les cas de hautes masses moléculaires (Tableau 17.1, Vollhardt p.634).Dans le groupement carbonyle, tant le carbone que loxygène sont hybridés en sp2 ce qui permet un recouvrement optimal des orbitales (Figure 17.1, Vollhardt p.633).
La différence délectronégativité entre le carbone et loxygène, de même que par la présence de la liaison
p entraînent la polarisation de la double liaison et un effet inductif-attractif sur les liaisons voisines Il résulte que le groupe carbonyle présente un moment dipolaire important (de lordre de 2.7 D) comparable à celui des halogénoalcanes.Ainsi, les premiers termes comme lacétone CH3COCH3 sont solubles dans leau, mais cette solubilité diminue rapidement lorsque le nombre datomes de carbone augmente et devient nulle à partir de 5 carbones.
La fonction carbonyle C=O possède une absorption caractéristique dans lU.V. entre 275 et 295 nm (
e 15 à 1100) due aux transitions de type p® p* et n® p* (Figures 17.6, Vollhardt p.637 et en haut de la p.637).Ils possèdent aussi une absorption très caractéristique dans linfrarouge entre 1 705 et 1 725 cm-1 pour les cétones et entre 1 720 et 1 740 cm-1 pour les aldéhydes (Figure 17.5 p.636).
Les protons aldéhydiques ont des déplacements caractéristiques en RMN-1H entre 9 et 10 ppm (Figure 17.3, Vollhardt p.635), alors que ceux des groupements méthyles adjacents aux cétones passent de 1,2 à 2,4 ppm.
Les spectres RMN-13C permet de distinguer aisément les aldéhydes et les cétones entre 170 et 200 ppm (Tableau 10.6, et Figure haut de la p.636) des alcènes qui eux résonnent vers 120 à 130 ppm.
8.1.3. Réactivité (Arnaud 18-2)
Les éléments structuraux qui déterminent la réactivité des aldéhydes et des cétones: lexistence dune liaison
p entre C et O, la présence de deux doublets libres sur O, et la différence délectronégativité entre C et O, entraînant la polarisation de la double liaison et un effet inductif-attractif sur les liaisons voisines.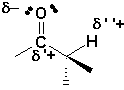
Ainsi, la présence de groupements électroattracteurs augmentera la charge positive du carbone carbonyle.
Les aldéhydes et les cétones démontrent une foule de réactions en tout genre. Toutefois, certaines réactions particulières valent tout particulièrement la peine dêtre mentionnées.
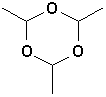
8.1.3.1. Acétals et hémiacétals (Vollhardt 17.7 et Arnaud 18.10)
Par chauffage en présence dun alcool et de HCl anhydre, les aldéhydes et les cétones se transforment en hémiacétals, puis en acétals:
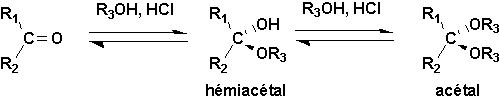
Cette réaction est réversible et très importante dans le cas des sucres comme nous le verrons plus loin.
De la même façon, les aldéhydes et les cétones se transforment en hémithioacétals, puis en thioacétals lorsquen présence de thiols:
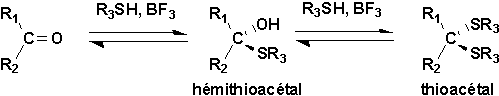
En présence damines, les aldéhydes et les cétones se transforment en hémiaminals, puis en imines:
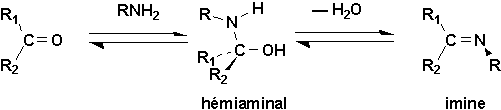
8.1.3.2. Oxydation et réduction (Vollhardt 17.4 et Arnaud 18.18)
Les aldéhydes soxydent pour donner un acide sans modification de la chaîne carbonée.
Cette oxydation est très facile, non seulement avec les oxydants conventionnels, mais aussi par loxygène de lair.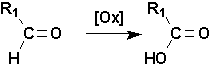
Les cétones soxydent beaucoup plus difficilement, et seulement en présence doxydants très énergiques
(KMnO4 concentré à chaud en milieu acide, H2CrO4, etc.). Cette oxydation coupe la chaîne entre le groupement carbonyle et un des carbones en a, avec formation de deux acides. Comme la coupure peut se produire des deux côtés, on obtient généralement un mélange dacides (4 au maximum si la cétone est dissymétrique).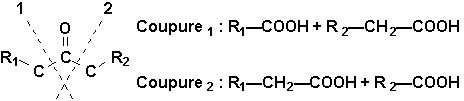
Certaines réactions de réduction à laide de métaux réduisent les cétones en diols du type pinacol (2,3-diméthylbutane-2,3-diol).
8.1.3.3. Polymérisation (Arnaud 18.19)
Le formaldéhyde, normalement gazeux existe aussi sous la forme dun trimère appelé trioxyméthylène ou trioxane, et dun polymère, le paraformaldéhyde. Ce sont des solides blancs pulvérulents (à létat de poudre fine) qui se dégradent facilement par action de la chaleur.
Lacétaldéhyde
existe sous la forme dun trimère, le paraldéhyde, un liquide dont le point débullition est 125°C, alors que lacétaldéhyde bout à 21°C. Ce trimère se décompose aussi facilement par chauffage en présence de traces dacide.8.1.4. État naturel (Arnaud 18-3)
Les cétones cycliques à grand cycle possèdent une odeur agréable et sont souvent utilisées en parfumerie. Les aldéhydes, spécialement ceux de faible poids moléculaire, possèdent une odeur piquante.
Les aldéhydes et les cétones sont fréquents dans les composés naturels, soit seuls ou accompagnant dautres fonctions. Citons en particulier les nombreuses essences végétales de la famille des terpènes.
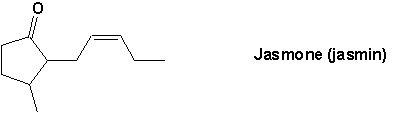

![]()
Citons aussi dautres exemples comme le camphre, la citronnelle, la vanilline et les divers stéroïdes.
Ils sont aussi utilisés dans lindustrie alimentaire: la butane-2,3-dione est le principal constituant odorant incorporé à la margarine. La muscone (3-méthylcyclopentadécanone), extraite des glandes odoriférantes du daim musqué mâle, est un parfum coûteux
8.1.5. Termes importants et utilisations (Arnaud 18-5)
Le formaldéhyde est laldéhyde industriellement le plus important. Il est spécifiquement obtenu par oxydation catalytique du méthanol par loxygène, et la production mondiale est denviron 9 millions de tonnes. Il est commercialisé sous la forme de solutions aqueuses à 37 % connues sous le nom de formol. Il est principalement utilisé dans la production de résines thermodurcissables. Nous le connaissons de mauvaise réputation pour la "célèbre" mousse isolante durée-formaldéhyde (la MIUF).
Lacétone
se prépare industriellement à partir du propène. Elle est utilisée principalement comme solvant (plus de 500,000 t/an aux USA) et entre dans la polymérisation du Plexiglass.La cyclohexanone entre elle dans la fabrication des nylons.
|
Encarts du chapitre 17 |
8.2 Énols et énones (Vollhardt chapitre 18)
8.2.1. Ions énolates (Vollhardt 18.1)
La labilité de lhydrogène en
a du groupement carbonyle amène une stabilisation de la forme énolate par résonance. Il en résulte des réactions de substitution de cet hydrogène labile. Les aldéhydes et cétones auront des pKa de lordre de 19-21.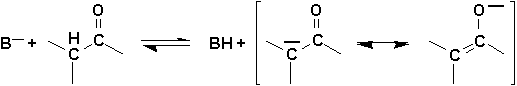
8.2.2. État naturel (Vollhardt problème 12, chapitre 18)
On retrouve beaucoup dénones et dénals dans la nature:
| hex-2-énal | arôme de tomates |
| oct-3-én-2-one | arôme de champignons |
| non-2-énal | goût de concombres |
| nona-2,4-diénal | parfum de concombres |
8.2.3. Tautomérie céto-énolique (Vollhardt 18.2)
La protonation dun ion énolate peut se produire soit sur loxygène, soit sur le carbone. Dans la réalité, on assiste toujours à un équilibre entre les deux formes, celle de la cétone, et celle de lénol.
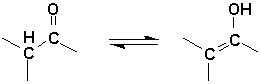
Cet équilibre est sensible à la catalyse acido-basique (souvent simultanée, quelques fois séquentielle):
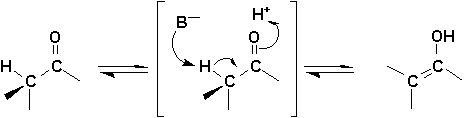
La position de cet équilibre dépend de plusieurs facteurs comme la structure, le solvant, la température, la concentration, ... Normalement, la forme céto est lespèce prédominante.
Ce sont 2 molécules différentes et non pas des formes de résonance.
Ce sont des tautomères, i.e. quelles sont facilement et rapidement inter convertibles dans les conditions ordinaires. Ainsi, le cyclohexanone existe sous deux formes tautomères, alors que le méthylidènecyclohexane et le 1-méthylcyclohexène sont des isomères structuraux.
La différence entre isomères structuraux et tautomères en est une de degré, et non despèce.
À léquilibre, pour les aldéhydes et cétones simples, la forme cétonique est favorisée. Par contre, dans le cas des fonctions 1,3-dicarbonylées, la forme énol est importante (à cause de la stabilisation par conjugaison entre >C=C< et >C=O et des ponts hydrogènes intramoléculaires).
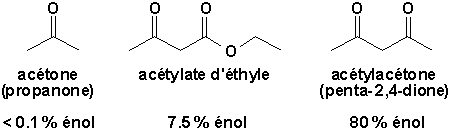
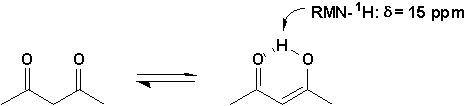
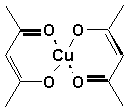
Les formes énoliques des composés 1,3-dicarbonylés prennent la forme dun cycle. On dit quils sont sous une forme chélatée (khêlê, grec, pince). Un chélate est un composé ou un atome métallique est pris en pince entre des atomes électronégatifs liés à un radical organique. Ex: acétylacétonate de cuivre.
Lexistence de cet équilibre céto-énolique interfère souvent avec le déroulement des réactions.
|
Encarts du chapitre 18 |
8.3 Les acides carboxyliques (Vollhardt chapitre 19)
Les acides carboxyliques ont comme formule générale
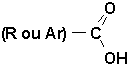
et doivent leur nom au groupe carboxyle
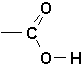
On les représente souvent par RCOOH ou RCO
2H.À cause de leurs doublets libres, les oxygènes sont basiques, alors que lhydrogène carboxylique est très acide. Le carbone porte une polarisation positive, et est considéré comme un électrophile.
8.3.1. Nomenclature des acides carboxyliques (Vollhardt 19.1 et Favre 5.2)
Tout comme les aldéhydes, les acides carboxyliques sont nommés daprès la nomenclature de substitution. On remplace le e de lhydrure parental par le suffixe oïque et on fait précéder le nom par le mot acide.
Étant donné quun acide carboxylique est toujours en bout de chaîne, il ne nécessite pas dindice de position, même lorsque lon parle de diacide. Il est à noter que le carbone dun groupement COOH est le numéro 1, ce qui fixe automatiquement la numérotation de la chaîne.
Lorsque lon rencontre des polyacides, on utilise le suffixe carboxylique. Lorsque le groupe COOH nest pas porté par la chaîne principale, on utilise le préfixe carboxy.
Il fut un temps où presque tous les acides gras (aliphatiques) portaient des noms triviaux. On les appelaient acides gras car, au début, ils étaient isolés des sources naturelles, les graisses. Aujourdhui, officiellement, il en reste six dans la série des acides saturés: les acides formique, acétique, propionique, butyrique, palmitique et stéarique. De plus, une différence majeure existe entre ces noms: un seul, lacide acétique, peut être utilisé comme hydrure parental fonctionnalisé, cest-à-dire peut engendrer des noms substitutifs comme par exemple "acide chloroacétique". Les cinq autres noms ne peuvent être utilisés que pour nommer lacide lui-même, non substitué: ainsi, le nom de:
Cl-CH2-CH2-COOH
sera acide 3-chloropropanoïque et non "acide 3-chloropropionique", encore moins "acide
b-chloropropionique".Lanion formé par le départ du proton acide porte le nom danion carboxylate, RCOO
-. Le nom est formé en remplaçant le suffixe oïque par ate et en retirant le mot acide.
Enfin, il est intéressant de noter les noms et sources naturelles des acides carboxyliques contenant de 1 à 6 atomes de carbone (Tableau 19.1, Vollhardt p.720). Ces noms triviaux provenaient de leur source dextraction, et non pas de leur structure.
8.3.2. Caractères Physiques (Vollhardt 19.2 et Arnaud 19-1)
Les acides acycliques linéaires sont des liquides ou des solides dont le point de fusion ne dépasse pas 100°C (lacide stéarique C17H35COOH dont sont faites les bougies fond à 70°C) (Tableau 19.2, Vollhardt p.723).
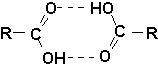
Par contre, ils possèdent les points débullition les plus élevés parmi les composés possédant une fonction simple sur une chaîne donnée. Ils présentent des liaisons hydrogène encore plus fortes que les alcools et se retrouvent souvent sous forme dun dimère cyclique.
Leur solubilité dans leau, totale jusquen C4, diminue et devient nulle à partir de C9. Les sels de sodium des acides de C12 à C18 forment ce que lon appelle les savons.
Les acides formique et acétique possèdent une odeur piquante et âcre de même quun goût aigre. Les acides de C4 à C8 possèdent une odeur exceptionnellement désagréable, mais par contre à faible concentration ils exaltent la senteur des parfums. Lacide pentanoïque (valérique) donne son odeur caractéristique au fromage bleu dAuvergne.
8.3.3. RMN des acides carboxyliques (Vollhardt 19.3)
Le proton carboxylique résonne à un champ extrêmement bas (
d = 10-13 ppm). Son déplacement chimique est fortement influencé par la concentration, la température, le solvant, à cause de la tendance marquée du groupement OH à participer à des liaisons hydrogène, tout comme les alcools (Figure 19.2, Vollhardt p.724).En ce qui concerne la RMN-13C, les valeurs de déplacement chimique du carbone carboxylique est similaire à ce qui est observé pour les aldéhydes et les cétones. Diverses structures de résonance explique ce phénomène (Figures p.725).
8.3.4. Réactivité (Arnaud 19-2)
Bien que le groupement carboxyle réunisse le groupement fonctionnel alcool et le groupement fonctionnel cétone, la réactivité nest pas la somme de la réactivité des deux groupements constituants. Il faut se souvenir que les deux groupes OH et C=O ne sont pas indépendants dans le groupe COOH et que le comportement de lun est fortement influencé par la présence de lautre. Il sont tous deux engagés dans une structure résonnante:
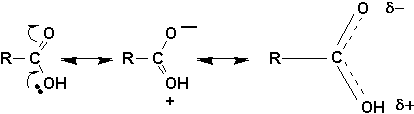
Ainsi,
| Groupe OH | Lhydrogène est plus labile que celui des alcools ou même des phénols. Les acides carboxyliques sont dissociés en solution aqueuse, surtout parce que la base conjuguée est stabilisée par résonance. |
| Groupe C=O | Le carbone fonctionnel des acides est nettement moins électrophile que celui des aldéhydes et cétones. En conséquence, les réactions daddition nucléophiles sont peu nombreuses. |
| Hydrogène en a | La labilité des hydrogène en a du carbonyle est beaucoup plus faible que pour les aldéhydes et les cétones, la forme énoliques étant inexistante. |
8.3.5. Propriétés acido-basiques (Vollhardt 19.4 et Arnaud 19-2)
Le caractère dominant des acides carboxyliques est ... leur acidité. Lorsque solubles dans leau la déprotonation seffectue rapidement et léquilibre suivant sétablit:
RCOOH + H2O ![]() RCOO
RCOO
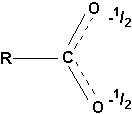
avec la forme de résonance suivante pour lanion carboxylate:
Ainsi, les acides carboxyliques donnent de nombreux "sels" lorsquils réagissent avec les métaux et les bases. Il va de soi que les groupements électroattracteurs vont exalter lacidité des acides carboxyliques et les rendre compétitifs envers les acides minéraux (Tableau 19.3, Vollhardt p.727).
8.3.6. État naturel (Arnaud 19-3)
De nombreux acides carboxyliques sont présents dans la nature, mais le plus souvent sous forme desters. Plus particulièrement, les corps gras végétaux ou animaux sont des esters du glycérol de divers acides gras acycliques à chaîne linéaire plus ou moins longues.
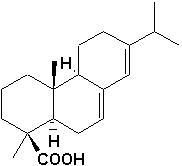
Un exemple dacide libre naturel est lacide abiétique (aussi appelé acide sylvique) contenu dans la résine de pin (® à droite).
On rencontre aussi lacide lactique dans le lait et les muscles
CH3CHOHCOOH
et lacide citrique dans les agrumes
HOOCCH2C(OH)(COOH)CH2COOH
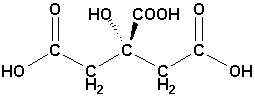
et divers acides aminés dans les protéines.
8.3.7. Termes importants et utilisations (Arnaud 19-5)
Lacide acétique est connu depuis très longtemps en raison de sa formation dans la transformation spontanée du vin en vinaigre. On le prépare par oxydation de lacétaldéhyde ou par synthèse à partir du méthanol et du monoxyde de carbone. Il est utilisé pour la préparation de tout ce que lon appelle acétates (de vinyle, déthyle, de cellulose).
Parmi les autres qui possèdent une grande utilisation industrielle, citons lacide acrylique, CH2=CHCOOH (esters polymérisables) et lacide stéarique, CH3(CH2)16COOH.
8.4 Diacides (Arnaud 20.15-20.19)
Parmi les diacides les plus important, citons notamment
lacide oxalique (éthanedioïque), HOOCCOOH, utilisé comme puissant réducteur
lacide adipique (hexanedioïque), HOOC(CH2)4COOH, qui est utilisé dans la synthèse du Nylon 6/6
lacide ortho-phtalique, qui permet dobtenir des colorants (les phtaléines) par réaction avec des phénols.
lacide para-phtalique (ou téréphtalique) sert à la fabrication du Tergal par polymérisation avec le glycol ordinaire.
8.5 Les dérivés des acides carboxyliques
On considère comme dérivant des acides les fonctions suivantes:
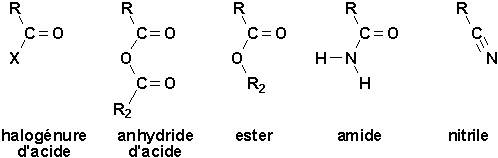
Ici, R et R2 peuvent être identiques ou différents. Ils peuvent aussi être remplacés par des groupes aryles, Ar.
Les quatre premiers dérivés contiennent tous la structure RCOZ, ou Z est ou contient un atome fortement électronégatif et porteur dau moins un doublet libre.
8.5.1. Halogénures dacides (Vollhardt 19.8, Arnaud 19.16 et Favre 5.3.1)
On les appelle aussi des halogénures dacyle ou halogénures dalcanoyle. Ils servent principalement dintermédiaires dans diverses réactions de synthèse.
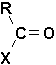
Ils proviennent du remplacement de groupe OH de la fonction carboxylique par un halogène, le plus souvent le chlore. On forme les noms en utilisant le nom du groupe acyle et en le faisant précéder de celui de lhalogénure approprié. On change la terminaison ique en yle ou oïque en oyle.
| CH3COC | chlorure dacétyl |
| BrOC(CH2)3COBr | dibromure de pentadioyle |
Ils sont insolubles dans leau, mais rapidement hydrolysés en acide carboxyliques
RCOCl + H2O ® RCOOH + HCl
Ils présentent de fortes interactions dipolaires, et en conséquence possèdent des points débullition plus élevés que les alcanes correspondants. Ils servent surtout à la protection de certaines fonctions lors de synthèses. Ils possèdent une odeur irritante et sont souvent utilisés comme gaz lacrymogènes.
Parmi les gaz de combat, le dichlorure de lacide carbonique ou phosgène, COCl2, fut responsable dun grand nombre de morts durant la première guerre mondiale. Son action toxique est due à son hydrolyse dans les poumons qui libère de lacide chlorhydrique. Le CCl4, qui est un des rares solvants organiques à pouvoir être employé dans les extincteurs, génère quand même un peu de phosgène à température élevée. Il en est de même avec le chloroforme. Le phosgène est un gaz mortel.
8.5.2. Anhydrides dacides et cétènes (Vollhardt 19.8, Allinger 8.15, Arnaud 19.17 et Favre 5.3.3)
On les appelle aussi tout simplement des anhydrides. Ils proviennent de la déshydratation intermoléculaire de deux molécules dacide:
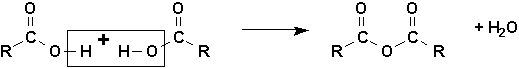
On les nomme en remplaçant le mot acide par le mot anhydride dans le nom dun acide carboxylique. Lorsque lanhydride est substitué, on utilise le préfixe "bis". On sen sert principalement pour effectuer les synthèses desters.
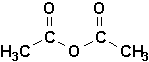
On connaît des anhydrides symétriques et mixtes: les premiers sont de loin les plus importants. Le plus connu est sans doute lanhydride acétique:
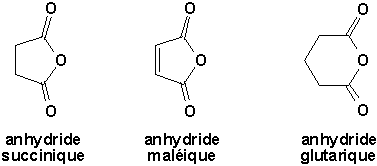
Les anhydrides cycliques sont très facilement formés à partir dacide dicarboxyliques, donnant ainsi des cycles à 5 ou 6 maillons:
Les substances qui dérivent dune molécule dacide par perte dune molécule deau sont des cétènes (du nom du composé parent CH2CO, le cétène qui dérive de lacide acétique):
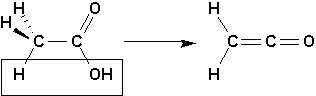
Les cétènes sont rarement isolés, on ne les voit apparaître que comme intermédiaires réactionnels.
8.5.3. Esters et lactones (Vollhardt 19.9, 20.4, Arnaud 19.18-20.25, Favre 5.3.2)
On parle depuis plusieurs chapitres des esters. On les rencontre principalement dans les réactions dhydrolyse et de saponification de divers produits naturels.
Rappelons ici que les alcools réagissent avec les acides carboxyliques pour former des esters. Cette réaction est catalysée par les acides forts comme H2SO4 et est réversible. On aboutit toujours à un état final ou les 4 produits cohabitent. Dans cet équilibre, cest le groupement OH de lacide qui part avec le H de lalcool.
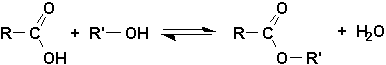
On a longtemps cru que lestérification était une neutralisation. Cest pourquoi on nomme les esters comme sils étaient des sels dalkyle. On change la terminaison ique en ate ou oïque en oate.
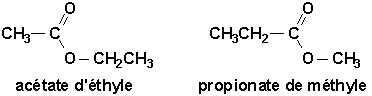
Lorsquils sont considérés comme substituants, nous utilisons le préfixe de forme R-oxycarbonyl-. Toutefois, certains noms ont la vie dure et vont continuer à être utilisés:
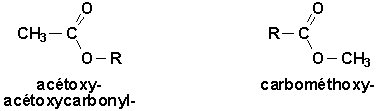
Le déplacement chimiques en RMN-1H sont les suivants:
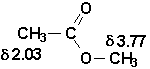
Il est important de mentionner que lon retrouve beaucoup desters en parfumerie et dans lindustrie alimentaire. Ils possèdent généralement une odeur fruitée ou florale.
Par exemple, les parfums de haute gamme contiennent parfois jusquà 100 substances différentes, alors que les moins coûteux peuvent nen contenir que quelques-uns. Larôme de café frais en contient plus de 500!
En alimentation, ce sont souvent les saveurs naturelles et artificielles ajoutées. Mentionnons ici lacétate disoamyle (odeur de banane), lisovalérate disoamyle (odeur de pomme) et le salicylate de méthyle (odeur de thé des bois) pour nen citer que quelques-uns.

Ils sont aussi utilisés dans la préparation de laques et de vernis. Les esters à point débullition élevé sont employés comme agents plastifiants pour les résines dont certaines sont elles-mêmes des polyesters.
On retrouve aussi les esters dans les glycérides (des esters de glycérol). Ici, R1, R2 et R3 sont des chaînes atteignant généralement entre 12 et 22 carbones.
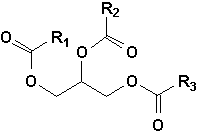
Les lactones sont des esters cycliques. On les nomme comme les hétérocycles quelles sont ou en utilisant le suffixe lactone et en remplaçant le lettre e finale de lhydrure parental par la lettre o:
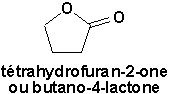
Il arrive encore parfois que la taille du cycle soit désignée par la lettre grecque correspondant au nombre de carbones séparant le C=O de lO dans le cycle: ainsi, la butano-4-lactone porte aussi le nom de
g-butyrolactone. Toutefois, cette nomenclature tend à disparaître.Les
g et d-lactones sont donc des cycles de 5 et 6 atomes, stables et sans contraintes très répandus dans la nature et particulièrement chez les végétaux. Les acides g et d-hydroxylés perdent spontanément une molécule deau et forment des lactones: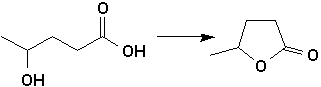
Cette réaction est très facile, au point quil est difficile disoler ces acides
g- et d-hydroxylés à létat pur à cause de léquilibre toujours présent. Par contre, les acides b-hydroxylés ne présentent pas cet équilibre car il comporteraient un cycle à 4 atomes trop tendu et instable.Les lactones sont généralement insolubles dans leau.
Les lactones macrocycliques forment le canevas fondamental de nombreux antibiotiques nommés des macrolides (Encart 19.2).
Enfin, il est intéressant de noter les analogues sulfurés des esters: les thionoesters.
8.5.4. Amides, lactames et imides (Vollhardt 19.10, 20.6, Allinger 8.12, Arnaud 19.19, Favre 5.5)
Les amides proviennent de la réaction dun acide carboxylique avec lammoniac ou une amine avec élimination dune molécule deau. Cette réaction est réversible. On rencontre 3 types damides, selon du degré de substitution de lazote:
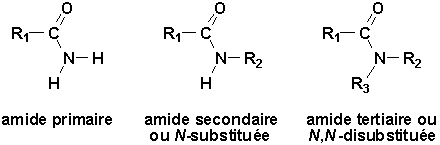
Les amides primaires sont solides, à lexception de la formamide (Fu 2.5°C). Les amides primaires et secondaires sont fortement associées par liaisons hydrogène intermoléculaires. La diméthylformamide, HCON(CH
3)2, est très polaire et constitue un excellent solvant des composés organiques et minéraux, tout en étant aprotique.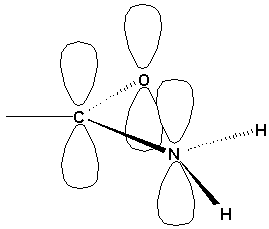
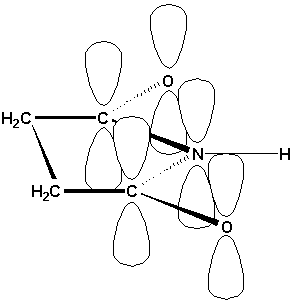
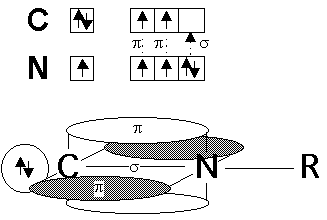
Dans lammoniac et les amines, lazote est hybridé en sp3 et possède une structure pyramidale. Dans les amides, lazote est hybridé en sp2 et est plan. Ainsi une énergie de conjugaison supplémentaire est obtenue par le recouvrement de lorbitale p de lazote contenant un doublet libre et lorbitale 2p du groupement carbonyle:
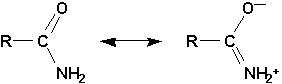
Du point de vue de la nomenclature, on nomme les amides à limage des acides carboxyliques. On change la terminaison ique en amide. Alternativement, on utilise le suffixe amide au nom de lhydrure parental avec lélision du e final. Lorsque lemploi dun préfixe est nécessaire, le terme carbamoyle est utilisé.
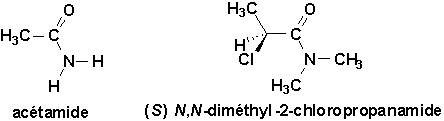
On sen sert principalement pour effectuer les synthèses.
La liaison CN dune amide simple possédant un caractère double prononcé, il ne pourra y avoir de rotation entièrement libre autour de cette liaison. Prenons lexemple de la N,N-diméthylformanide
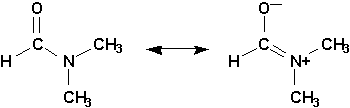
Il en résulte que les deux groupements CH3 sont magnétiquement différents, lun est en cis, lautre est en trans de lhydrogène porté par le carbone. En RMN-1H, ils donnent deux pics distincts,
d 2.97 ppm pour le groupe près de loxygène et d 2.88 ppm pour celui près de lhydrogène. Lorsque la température sélève, les deux pics sélargissent, et à 111°C les deux pics coalescent en un pic unique qui samincit si on élève encore la température, la rotation rapide autour de la liaison CN étant facilitée par laccroissement dénergie.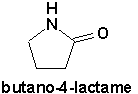
Tout comme pour les lactones, la formation dune amide par cyclisation dun acide aminocarboxylé donne un lactame.
Ils sont nommés de la même façon que les lactones.
Les imides contiennent deux groupements acyles liés au même azote. Le groupement qui les caractérise est CONHCO. Les imides cycliques sont les plus importantes. On les nomme comme des diacylamines ou en utilisant le terme imide.
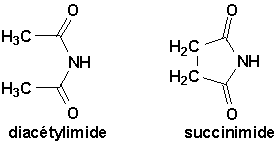
Dans la succinimide, lazote est hybridé en sp2, ce qui confère un intéressant système
p à la molécule (® à droite):Les dérivés de lurée et les uréthanes
sont des composés nommés par filiation avec les plus simples dentre eux, respectivement lurée et luréthane. Ce sont des dérivés très stables de lacide carbonique et de lacide carbamique (instables).| Acide carbonique | HOCOOH |
| Acide carbamique | H2NCOOH |
| Urée | H2NCONH2 |
| Uréthane | H2NCOCH2CH3 |
| Un uréthane | RNHCOR |
8.6 Autres groupes fonctionnels contenant des liaisons multiples carbone-azote
8.6.1. Nitriles et analogues (Vollhardt 20.8, Allinger 10.1 et Favre 5.5.3)
Le premier terme de la série des nitriles, le cyanure dhydrogène, HCº N (improprement appelé acide cyanhydrique) possède un caractère acide qui le différencie nettement des autres termes de la série. Ici, le carbone et lazote sont tous deux hybridés en sp et le doublet libre de lazote occupe une de ces deux orbitales sp. La structure électronique est semblable à celle de léthyne.
On nomme les nitriles comme les amides: en remplaçant la terminaison ique par nitrile ou en utilisant simplement le suffixe nitrile. Lorsque lemploi dun préfixe est nécessaire, le terme cyano est utilisé.
| acétonitrile | CH3CN |
Les nitriles possèdent un moment dipolaire élevé; leur point débullition est légèrement plus élevé que les alcools de masse moléculaire comparable [Note].
On se sert principalement des nitriles pour effectuer les synthèses.
En IR, la vibration C+N apparaît vers 1250 cm-1, i.e. dans la même zone que la vibration Cº C. En RMN-1H, les protons voisins du groupe nitrile sont quasiment aussi déblindés que ceux dautres dérivés des acides carboxyliques et des alcynes (Tableau 20.3, Vollhardt p.793). Labsorption en RMN-13C du carbone du groupe nitrile apparaît vers 112 à 126 ppm, alors que les alcynes apparaissaient vers 65 à 85 ppm.
Les groupes suivants sont des analogues des cyanures:
| isocyanure | NºC: |
| cyanate | O-CºN |
| isocyanate | N=C=O |
| fulminate | ON=C |
| carbodiimide | N=C=N |
Les isocyanures sont remarquables par leur structure peu courante et également par leur odeur extrêmement désagréable. Ils constituent, comme loxyde de carbone CO, lun des rares composés stables renfermant un atome de carbone divalent, stabilisé par un nombre important de formes mésomères chargées.
Le doublet délectron du carbone constitue le site réactionnel. La liaison de coordinance implique quil ny a aucune charge présente dans la molécule.
8.6.2. Hydrazones et oximes (Allinger 10.1)
Les hydrazones, R2C=NNH2, et les oximes, R2C=NOH, sont des imines dérivant de lhydrazine, NH2NH2, et de lhydroxylamine, NH2OH respectivement. Ce sont en général des dérivés cristallisés stables. Latome dazote des imines est hybridé en sp2 et lisomérie géométrique peut être observée pour la liaison double carbone-azote. Latome dazote existant dans les imines ne subit pas une inversion aussi aisée que dans le cas de lammoniac et des amines.
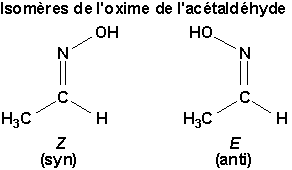
8.6.3. Groupes nitro et analogues (Allinger 10.2)
Le groupe nitro, NO2, est isoélectronique de lanion carboxylate, CO2- et peut être écrit sous deux formes de résonance équivalentes:
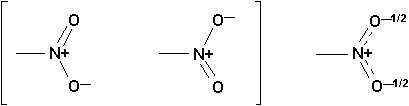
Le groupe nitro est fortement électronégatif et électroattracteur et les dérivés nitrés sont des substances à point débullition élevé, mais, contre toute attente, ils sont peu solubles dans leau.
Le groupe nitro est capable de stabiliser dune manière très efficace une charge négative sur un atome adjacent.
Du point de vue de la nomenclature, le groupe nitro est toujours nommé comme un préfixe.
Les dérivés, les nitrates (esters de lacide nitrique) et les nitramines (N-nitroamines) sont des explosifs puissants qui détonnent au choc.
Les dérivés contenant le groupe nitroso, N=O, sont plutôt rares. Les dérivés nitrosés, ou le groupe nitroso est porté par un carbone primaire ou secondaire sont instables. Ils se dimérisent ou se tautomérisent en oximes.
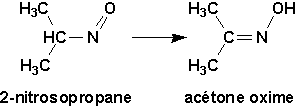
Du point de vue de la nomenclature, le groupe nitroso est toujours nommé comme un préfixe.
Enfin, les composés de structure ON=O sont les nitrites (esters de lacide nitreux).
8.6.4. Dérivés azoïques, diazoïques et azotures (Allinger 10.3, Arnaud 17.20)
La triple liaison azote-azote de la molécule N2, Nº N, est la plus forte liaison covalente connue (226 kcalmol-1). Il nest donc pas surprenant que la plupart des composés renfermant des liaisons doubles azote-azote tendent à se décomposer ou à réagir de manière telle quil y ait libération dune molécule dazote.
Les dérivés azoïques contiennent le groupement N=N. Ceux de structure RN=NH sont extrêmement instables et se décomposent rapidement en RH et N
2, alors que la stabilité des dérivés disubstitués RN=NR est bien supérieure et dépend de la nature du groupe R. Ainsi, lazométhane, CH3N=NCH3 est un liquide jaune qui est stable jusquà environ 400 °C.Le plus important des dérivés diazoïques est le premier terme: le diazométhane (gaz jaune, Eb: -23°C):
![]()
Cest un des réactifs les plus intéressants et les plus utilisés de la chimie organique en dépit du fait quil soit hautement explosif, instable à température ambiante, extrêmement toxique et préparé exclusivement à partir de produits provoquant de dangereuses réactions allergiques.
Du point de vue de la nomenclature, les dérivés diazoïques sont toujours nommé comme un préfixe en utilisant le terme diazo.
Les composés diazoïques sont très utilisés dans lindustrie des colorants. De plus, la protonation du groupement diazoïque change la conjugaison de la molécule, de telle sorte quils sont aussi souvent utilisés comme indicateurs de pH. Citons notamment le Méthylorange et le Rouge Congo (section 22.11 p.896-7).
Enfin, les azotures portent un groupement N3. Ce sont des dérivés organiques de lacide hydrazoïque, HN3, qui est très toxique et instable. Les azotures de faible masse moléculaire sont souvent explosifs (sacs gonflables). La structure du groupement azido (tel quil est utilisé comme préfixe) est la suivante:
![]()
|
Les étudiants doivent lire la section 19.14 dans Vollhardt
p.749-753 |
8.7 Composés dicarbonylés et autres composés bifonctionnels (Vollhardt 23, Arnaud 20)
Il sagit de dialdéhydes, de dicétones et daldéhydes-cétones, dont les deux fonctions peuvent occuper des positions relatives diverses.
Nous ne verrons ici que deux catégories de ces composés dicarbonylés.
8.7.1. Quinones (Vollhardt 22.8-22.9)
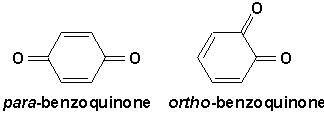
Les quinones sont des dicétones éthylénique conjuguées cycliques aussi appelées des cyclohexadiènediones ou benzoquinones (la nomenclature des quinones est décrite dans Favre aux pages 77 et 78.) telles que:
Toutes les liaisons doubles étant conjuguées, certaines formes de résonance les apparentent aux composés benzéniques:
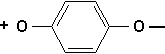
On observe les quinones comme étant le résultat dune réaction de réduction sur un paradiphénol. Les quinones sont des composés colorés absorbant dans le domaine du visible du spectre. Beaucoup de ces structures sont formées dans la structure de la lignine lors du jaunissement du papier.
Beaucoup de quinones naturelles servent dagents doxydation réversibles. Citons entre autre les ubiquinones, présentes dans la vitamine K
1, et les hydroquinones utilisées comme révélateurs photographiques (section 22.9).8.7.2. Acides-alcools (Arnaud 20.23-20.25)
Les acides
a-alcools, comme lacide lactiqueCH
3CHOHCOOHpeuvent donner lieu à une estérification réciproque de deux molécules avec formation dun diester cyclique, appelé un lactide
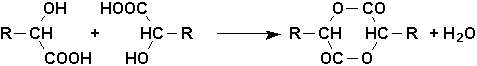
Cette réaction est si facile quil nest pas possible de conserver un acide
a-alcool, si ce nest sous forme dun de ses sels.Les acides
g- et d-alcools ont leurs deux fonctions suffisamment proches dans lespace pour quil puisse se produire une estérification intramoléculaire, donnant une g- ou une d-lactone comme nous lavons vu précédemment.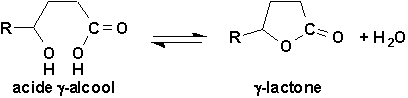
Faire les exercices de nomenclature qui sont présentés dans Favre
Autres exemples: (réponses après les structures)
a) 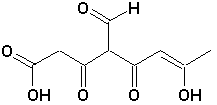 |
b) CH3COCl |
c) 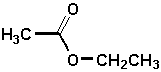 |
d) BrOC(CH2)3COBr |
e) 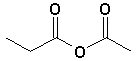 |
f) CH3CH2COOCOCH3 |
g) 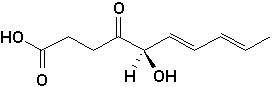 |
h) 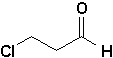 |
i) 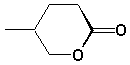 |
j) 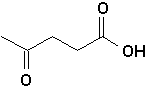 |
Réponses:
a) acide (Z)-4-formyl-7-hydroxy-3,5-dioxooct-6-énoïque
b) chlorure dacétyle
c) acétate déthyle
d) dibromure de pentanedioyle
e) anhydride acétique-propionique
f) anhydride acétique-propionique
g) acide (5R,6E,8E)-5-hydroxy-4-oxodéca-6,8-diénoïque
h) 3-chloropropanal, ou b-chloropropanal
i) hexano-5-lactone ou d-hexanolactone
j) acide 4-oxopentanoïque
| Retour au menu |
|
ABCSITE © copyright 2000 |