abcsite.free.fr
![]()
Chimie Organique
|
|
5. Groupements fonctionnels à liaison simple
| Dans ce chapitre | |
| 5.1. | Acides et bases |
| 5.2. | Dérivés halogénés, les halogénoalcanes |
| Modèles Moléculaires en format .pdb: Composés Halogénés | |
| 5.3. | Alcools, éthers et thiols |
| Modèles Moléculaires en format .pdb: Alcools | |
| Modèles Moléculaires en format .pdb: Éthers | |
| 5.4. | Amines |
| Modèles Moléculaires en format .pdb: Amines | |
| 5.5. | Organophosphorés |
| 5.6. | Organosilicés |
| 5.7. | Organoboriques |
| 5.8. | Organométalliques |
| 5.9. | Exercices de nomenclature |
Les groupements fonctionnels simples comporteront au moins un hétéroatome (X, O, S, N). De plus, ce groupement est lié au carbone de façon covalente par une liaison simple, s.
Il faut aussi tenir compte des hybridations des divers atomes impliqués. Rappelons que les principales hybridations rencontrées sont:
sp angle de 180°, molécule linéaire
sp2 angles de 120°, forme un triangle
sp3 angles de 109°, forme un tétraèdre
dsp3 angles de 90° et 120°, forme une bipyramide à base triangulaire
d2sp3 angles de 90°, forme un octaèdre
Rappelons aussi que les divers facteurs qui vont influencer les formes géométriques seront:
La distribution dissymétrique (ex: CH2F2) qui formera des liaisons polaires
La tension des cycles
Les contraintes stériques (ex: groupements tert-butyles)
Nous allons tout dabord effectuer une brève révision des concepts dacides et de bases.
5.1. Acides et bases (Vollhardt 6.7)
En 1887, Svante Arrhénius [Note] décrivait ainsi le concept dacide et de base:
"Un acide est une substance qui contient un excès de H+, alors quune base est une substance contenant un excès de OH-".
Il est peu vraisemblable quun proton existe ainsi à létat isolé en solution aqueuse. En effet, le rapport:
H· (H2O)n+ / H+= 10190
est supérieur au nombre datomes de lunivers (» 1097)!
Le concept dArrhénius, profond et brillant pour son époque, ne sapplique de plus quaux solutions aqueuses. Cest ce qui a amené Brønsted et Lowry à proposer en 1923 un autre concept:
"Les acides sont des donneurs de protons, alors que les bases sont des accepteurs de protons".
HCl + H2O ![]() H3O+ + Cl-
H3O+ + Cl-
Ceci a mené au concept dacides et de bases conjuguées. Lorsque lacide conjugué est un acide fort, sa base conjuguée est une base faible et vice versa.
HA (acide conjugué) ![]() A- (base conjuguée) + H+
A- (base conjuguée) + H+
Un corollaire important de lidée de Brønsted et Lowry est le fait quà léquilibre, tout réaction de transfert de proton favorisera la formation de lacide le plus faible et de la base la plus faible au dépens de la base la plus forte et de lacide le plus fort. Cette définition, pour large quelle soit, était limitée par lemploi du mot proton (cest ce que lon appelle le culte du proton).
En 1923, Lewis proposa de définir:
"Un acide est un accepteur de doublet électronique, et une base est un donneur de doublet électronique".
Dans la théorie de Lewis, une réaction acide-base est un partage du doublet électronique dune base avec un acide. Le tout pouvant aussi bien former une liaison covalente. Ainsi, le transfert du proton ne devient quun cas particulier de cette réaction.
Ainsi, selon la définition de Lewis, les acides comprennent:
Les ions positifs:
Acide base Complexe de coordination
Ag+ + 2 NH3 ® [ NH3AgNH3 ] +
NO2+ + CH2=CH2 ® +CH2CH2NO2
CH3+ + CH3OCH3 ® (CH3)3O+
Les composés présentants une lacune électronique (octet incomplet)
Acide base Complexe de coordination
AlCl3 + (CH3)3CCl ® AlCl4 + (CH3)3C+
Rappel de définitions:
HA + H2O ![]() H3O+ + A
H3O+ + A
Ka = 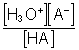 = 10-pKa
= 10-pKa
pKa = - log Ka
= ![]()
Ka x Kb = Kw = 10-14
pKa + pKb = 14
pKa = pH dune solution à demi neutralisée
Passons maintenant à la description des diverses classes de composés à fonction simples.
5.2. Dérivés halogénés, les halogénoalcanes (Vollhardt 6.1-6.2)
Aussi appelés halogénures, ces composés résultent du remplacement dun atome dhydrogène par un ou plusieurs halogènes, principalement Cl, Br, I, et plus rarement F.
Nous parlerons principalement ici des dérivés monohalogénés, RX.
Les halogénures seront toujours nommés comme préfixes des hydrures parentaux. En cas de disparités, le texte de Favre prime TOUJOUR sur Vollhardt.
5.2.1. Caractères Physiques (Vollhardt 6.2 et Arnaud 13-2)
Les dérivés halogénés possèdent des points débullition plus élevés que ceux des hydrocarbures correspondants. Il ne faut pas oublier que le point débullition augmente avec la masse moléculaire. Ils ont également une densité supérieure, principalement les dérivés polyhalogénés bromés et iodés (ex: r CH3I = 2,28, r CHBr3 = 2,85). Se référer au Tableau 6.2 (Vollhardt p.178).
Ils sont insolubles dans leau, mais sont eux-mêmes des bons solvants pour de nombreux composés organiques, notamment les corps gras. On na quà penser ici au chloroforme, CHCl3, et au dichlorométhane, CH2Cl2.
La liaison CX étant polarisée
, les dérivés halogénés possèdent donc un moment dipolaire moléculaire non nul (à moins que la géométrie annule les vecteurs). Les interactions de London sont aussi plus fortes dans le cas des halogènes les plus lourds, parce que les électrons sont moins maintenus par les noyaux atomiques. Ce phénomène porte le nom de polarisabilité. Cest une mesure de la capacité des électrons de la couche de valence à suivre linfluence dun champ électrique externe.Dailleurs, la force de la liaison CX diminue, et la longueur du lien augmente, lorsque lon descend dans le tableau périodique. Cette diminution de lénergie de liaison est principalement due au manque defficacité du recouvrement des orbitales qui est maximal entre des orbitales de même nombre quantique principal (n).
Les dérivés halogénés sont souvent fortement odorants.
| On saute le reste du chapitre 6 de même que le chapitre 7 |
5.2.2. Réactivité (Arnaud 13-2)
Leur réactivité est essentiellement due aux perturbations causées par la forte électronégativité de lhalogène. Le carbone se trouve porteur dune charge partielle positive, il devient donc la cible de lattaque des réactifs nucléophiles.
Plus précisément, les dérivés halogénés se prêtent à deux types de réaction: substitution et élimination. Ces mécanismes feront lobjet du cours de chimie organique II.
5.2.3. Dérivés fluorés (Arnaud 13-4)
Les dérivés fluorés constituent une classe très particulière des dérivés halogénés. Contrairement à tous les autres dérivés halogénés, les dérivés fluorés sont chimiquement totalement inertes. Ceci est du à la très faible polarisabilité de la liaison CF, elle-même une conséquence de la petitesse de latome de fluor.
Ils ne peuvent être préparés par action directe du fluor sur les hydrocarbures car la réaction est très violente et difficile à contrôler.
Une réaction particulière permet toutefois dobtenir le tétrafluoroéthylène, en vue de sa polymérisation ultérieure:
2 CHClF2 ![]() F2C=CF2 + 2 HCl
F2C=CF2 + 2 HCl
5.2.4. Termes importants et utilisations (Arnaud 13-5)
Les dérivés halogénés trouvent dinnombrables applications. Citons entre autres:
Intermédiaires de synthèse
Solvants (CHCl3CH2Cl2), fluide dextincteur (CF3Br), fluides frigorifiques (Fréons), fluides propulseurs dans les bombes aérosols (les fameux chlorofluorocarbones ou CFC, tel le Fréon-12, CF2Cl2). On pense même pouvoir utiliser les dérivés perfluorés comme sang artificiel!
Les dérivés polychlorés du biphényle (les fameux biphényles polychlorés, BPC) sont des liquides utilisés comme fluides de refroidissement et disolement dans les transformateurs.
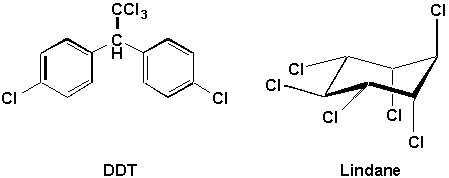
Insecticides, pesticides (comme le dichlorodiphényltrichloroéthane, D.D.T., et le 1,2,3,4,5,6-hexachlorohexane, HCH ou Lindane) et herbicides (chlorophénols).
Les polymères tel les chlorures de polyvinyle (PVC) et les polychloroprènes (caoutchouc synthétiques).
Le Téflon, qui est un polytétrafluoroéthylène, et qui possède de remarquables propriétés de résistance chimiques, mécaniques et thermiques. On na quà penser aux poêles Téfal, aux engrenages auto lubrifiés et aux isolants de type Gore-Tex.
Toutefois, aussi utiles soient-ils, ils ne faut pas oublier que nombre dentre eux créent dénormes problèmes environnementaux (pesticides, fréons, BPC).
Ainsi, on retrouve dans les pesticides des molécules comme de D.D.T. (dichlorodiphényletrichloroéthane) et le Lindane.
| Les problèmes 1 à 5 des pages 201 à 205 peuvent être faits. |
5.3. Alcools, éthers et thiols (Vollhardt chapitres 8 et 9)
Les alcools sont des composés dans lesquels un groupement hydroxyle est lié à un carbone saturé (sp3). Leur formule est ROH.
Si le groupement OH est lié à un cycle benzénique, il sagit dun phénol.
Si le groupement OH est lié à un carbone appartenant à une double liaison, il sagit dun énol.
On distingue 3 classes dalcools en fonction du degré de substitution du carbone qui porte la fonction:
- Alcool primaire, RCH2OH
- Alcool secondaire, RRCHOH
- Alcool tertiaire, RRRCOH
Les alcools seront nommés en changeant la dessinence -e des alcanes par la dessinence -ol. Toutefois, en présence dun groupement fonctionnel plus important, ils seront nommés comme préfixes des hydrures parentaux en tant que groupement hydroxy. Le texte de Favre prime sur la section 8.1 de Vollhardt.
5.3.1. Structure et propriétés physiques (Vollhardt 8.2 et Arnaud 15-1)
La Figure 8.1 (Vollhardt p.241) présente la structure tétraédrique de loxygène. La structure est déformée par rapport à un tétraèdre régulier parce que les doublets libres occupent un volume plus grand que les électrons liants. De plus, la forte électronégativité de loxygène diminue la longueur des liaisons et polarise la molécule, comme démontré dans la Figure au bas de la p. 241 (Vollhardt).
Aucun alcool nest gazeux à température de la pièce. Les alcools acycliques sont liquides jusquen C12, solides au-delà.
Les points débullition des alcools sont beaucoup plus élevés que ceux des hydrocarbures correspondants, et de presque tous les autres composés fonctionnels portant le même nombre datomes de carbone (Tableau 8.1, Vollhardt p.271).
Ce nest pas seulement du à laugmentation du poids moléculaire. Par exemple, prenons les points débullition suivants:
| CH3CH3 | - 88,5 °C |
| CH3OCH3 | - 24,0 °C |
| CH3CH2OH | + 78,3 °C |
On peut voir que le point débullition grimpe énormément. Cela est du à lexistence de liaisons intermoléculaires que lon appelle liaisons hydrogène. Elles résultent de la forte polarisation de la liaison OH qui entraîne la présence de charges partielles sur O (d) et H (d+). Cela permet des associations diverses telles (Figure 8.2 Vollhardt p.242):
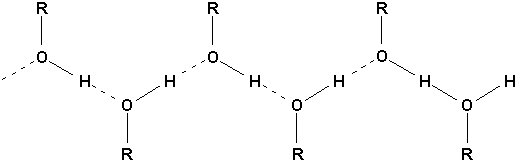
Cest cette attraction supplémentaire quil faut vaincre pour faire évaporer les molécules. Bien que beaucoup moins forte quune liaison covalente, la liaison hydrogène représente quand même une importante barrière énergétique à franchir. La liaison hydrogène est présente partout ou lon rencontre une fonction OH ou NH. Elle résulte toujours en une augmentation de lattraction des molécules.
Étant donné que les chaînes hydrocarbonées sont par nature hydrophobes (hydro, grec, eau; phobos, grec, peur), celles-ci ne se dissolvent pas dans leau. Par contre, la présence dun groupement alcool capable de faire des liaisons hydrogènes rend la molécule hydrophile. La solubilité dans leau des alcools dépendra donc de la balance hydrophobe-hydrophile des molécules (Figure 8.3, Vollhardt p.243).
Les alcools sont toxiques. Labsorption de méthanol peut provoquer la cécité. Léthanol possède des propriétés de vasodilatation importantes, de même quil inhibe laction de certains neurotransmetteurs.
5.3.2. Propriétés acido-basiques (Vollhardt 8.3)
Les alcools possèdent deux caractéristiques réactionnelles importantes:
Les liaisons CO et OH sont grandement polarisées à cause de la grande électronégativité de loxygène.
Loxygène possède deux doublets délectrons libres.
Les alcools sont légèrement acides (Tableau 8.2, Vollhardt p.244) et engendrent, à la suite de leur déprotonation à laide dune base forte, des sels appelés alkoxydes ou alcoolates qui sont des nucléophiles puissants.
ROH + B- ®R-O- + BH
CH3CH2ONa = éthanolate de sodium
De plus, lencombrement stérique, la polarisabilité et les effets inductifs contrôlent les valeurs de pKa des alcools. Lordre général des pKa des alcools en solution est:
CH3OH < primaire < secondaire < tertiaire
De plus, leffet inductif des groupement fortement électroattracteurs (Tableau 8.2, colonne de droite) a proximité du groupement -OH augmente lacidité de celui-ci.
Dun autre côté, les paires délectrons libres de loxygène rendent les alcools basiques.
Il en résulte que les alcools sont ce que lon appelle des ampholytes (amphos , grec, les deux), i.e. des molécules qui sont à la fois des acides et des bases. On dit que le groupement -OH est un groupement amphotère.
5.3.3. Oxydation (Arnaud 15.8)
En présence des oxydants usuels (KMnO4, ou K2CrO7/H2SO4), les alcools subissent une oxydation selon le schéma suivant:
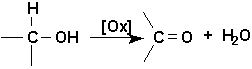
Les chemins doxydation des trois types dalcools sont les suivants:
Alcools primaires
RCH2OH ® RCHO (aldéhyde) ® RCOOH
Alcools secondaires
RCHOHR ® RCOR (cétone) ® RCOOH + RCOOH
Alcools tertiaires
RC(R)(R)OH ® Alcène ® Produits doxydation
| Encarts du chapitre 8. On saute le reste du chapitre 8. Notions importantes 1 à 4 dans Vollhardt à la page 270. Les problèmes 1 à 9 des pages 270 à 276 peuvent être faits. |
5.3.4. Diols et polyols (Arnaud 20.7 à 20.11)
Ces composés réunissent dans une même molécule deux fonctions alcool, qui peuvent être séparément primaires, secondaires ou tertiaires.
Les deux fonctions OH dun diol ne peuvent pas se retrouver sur le même carbone, car il sagirait alors dun hydrate daldéhyde ou dune cétone instable.
5.3.5. Diols (glycols) (Arnaud 20.8 à 20.10)
Les caractères physiques des diols sont différents de ceux des alcools simples, tout simplement parce quil y a plus dun groupement OH.
À cause de laugmentation du nombre de liaisons hydrogène, on observe:
une augmentation notable du point débullition
CH3CH2OH; Eb = 78,3°C
HOH2CCH2OH; Eb = 198°C
une augmentation de la viscosité
un accroissement de la solubilité dans leau
une saveur sucrée
Pour les triols et les polyols, ces caractères sont encore plus accentués.
Du point de vue de la réactivité, les propriétés des diols sont dans lensemble celles des alcools répétées deux fois (par exemple: formation de diesters). Cependant certaines propriétés particulières émergent selon la position particulière des 2 fonctions -OH.
Ainsi, les a-diols se déshydratent en réarrangeant la molécule et formant une cétone (mais si lune des deux fonction est un alcool primaire, le résultat est un aldéhyde):
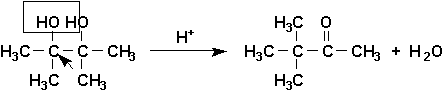
Un des diols les plus important est léthanediol, HOH2CCH2OH, encore appelé glycol ordinaire ou éthylène-glycol (improprement). Il est notamment employé dans la fabrication dexplosifs (esters nitriques) et comme antigel.
5.3.6. Triols (Arnaud 20.11)
Un seul triol est vraiment important, le propane-1,2,3-triol, ou glycérol (improprement appelé glycérine). On le retrouve à létat naturel sous forme de triester
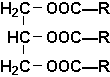
qui constituent les corps gras, graisses ou huiles, dorigine animale ou végétale (huile de lin, dolive, darachide, beurre, etc.). Les acides RCOOH sont essentiellement des acides acycliques linéaires à longue chaîne, appelés précisément acides gras.
Lorsque lon hydrolyse (H2O) ces corps gras, on obtient le glycérol et les acides gras à létat libre. Par contre lors dune saponification (OH), ces acides gras sont transformés en sels de sodium ou de potassium qui constituent les savons.
Le glycérol est un liquide très visqueux, bouillant à 290°C, de saveur sucrée et toxique.
Ces principaux dérivés sont des esters:
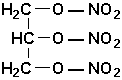
avec HNO3 on obtient le trinitrate de glycérol, qui est un explosif (improprement appelé nitroglycérine)
Mélangé avec une matière inerte pour en réduire sa sensibilité au choc, il constitue la dynamite, inventée par Alfred Nobel.
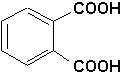
avec lacide ortho-phtalique
il se produit une réaction de polyestérification qui conduit à des résines thermodurcissables (appelées résines glycérophtaliques).
5.3.7. Polyols (Arnaud 20.11)
Les seuls polyols importants contiennent en outre une fonction carbonylée; ce sont les sucres qui seront étudiés plus loin.
Effectuons toutefois lexercice de nomenclature suivant:
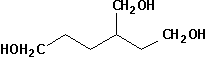
3-(hydroxyméthyl)hexane-1,6-diol
5.3.8. État naturel (Arnaud 15.10)
Il existe divers alcools dans la nature, spécialement les alcools terpéniques végétaux comme le menthol, le terpinéol, le linalol. Dans les organismes animaux, on retrouve notamment le cholestérol et la vitamine A. Mais le plus souvent, dans la nature, elle est associée à dautres fonctions et est souvent estérifiée.
5.3.9. Termes importants et utilisations (Arnaud 15.20)
Pour le tonnage produit et la diversité des utilisation, le méthanol est le plus important des alcools. On lobtient industriellement par synthèse à partir dun mélange ce monoxyde de carbone et dhydrogène:
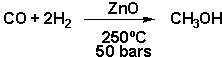
La production mondiale annuelle de méthanol est environ de 13 millions de tonnes. Le méthanol sert aussi à la synthèse de lacide acétique, ainsi que du métacrylate de méthyle (H2C=CH(CH3)COOCH3) dont la polymérisation donne le Plexiglass.
On produit environ 1 million de tonnes déthanol par an, soit par hydratation de léthylène, soit par fermentation de solutions sucrées naturelles (raison, betterave) ou artificielles (hydrolyse de lamidon ou de la cellulose).
Le propan-2-ol est la matière première de la préparation de lacétone. Et divers alcools comportant de 6 à 16 carbones sont utilisés dans la fabrication des détergents.
5.3.10. Éthers (Vollhardt 9.5 et Arnaud 15.21)
Les éthers sont caractérisés par lenchaînement fonctionnel suivant:
ROR
Du point de vue de la nomenclature, les éthers sont généralement des alkyloxyalcanes (Favre chapitre 4.2), les quatre premiers termes faisant toujours exception. Ainsi, on rencontre les groupes suivants:
| méthoxy | -OCH3 |
| éthoxy | -OCH2CH3 |
| propoxy | -OCH2CH2CH3 |
| butoxy | -OCH2CH2CH2CH3 |
| pentyloxy, hexyloxy, ... | |
Ainsi, la molécule CH3CH2-O-CH3 sappellera-t-elle le méthoxyéthane, et la molécule CH3-O-CH2CH2-O-CH3 le 1,2-diméthoxyéthane. Les préfixes di, tri, etc. sont utilisés lorsque les groupes sont contractés simples. Lorsque ces groupes sont non contractés et complexes, on utilise les préfixes bis, tris, etc.
Toutefois, on commence souvent à utiliser la nomenclature par remplacement lorsque les noms deviennent encombrants et complexes. Un tel exemple est donné pour le tétraglyme dans Favre p.32.
Il faut toutefois se rappeler que lon nutilise jamais le terme éther dans la nomenclature de ceux-ci.
On utilise aussi parfois le terme "oxyde":méthoxyéthane ou CH3-O-CH2CH3
oxyde déthyle et de méthyle
On peut aussi considérer les époxydes comme des éthers internes obtenus par déshydratation des a-diols.
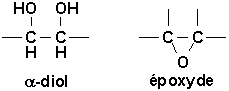
En ce qui concerne leurs propriétés physiques, labsence de possibilités de liaisons hydrogène entraîne de fortes diminutions des points débullition et de leur solubilité dans leau en comparaison avec les alcools dont ils sont les isomères (tableau 9.1, Vollhardt p.291).
Ils sont chimiquement très peu réactifs et, pour cette raison, sont souvent employés comme solvants. Des éthers couramment utilisés sont présentés au haut de la p.291 dans Vollhardt. Tous ces éthers ont reçus des noms consacrés par lusage qui seront difficile à éliminer.
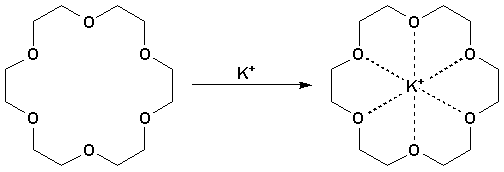
La présence des 2 doublets libres rend toutefois les éthers apte à former des liaisons de coordinence avec les molécules ayant des orbitales vides (BF3, organomagnésiens), ou soit avec des cations (éther couronnes). La figure suivante montre la structure dun polyéther couronne fréquemment utilisé, léther-18-couronne-6 (1,4,7,10,13, 16-hexaoxacyclooctadécane). Les éther couronnes montrent la remarquable aptitude à lier fortement les cations métalliques, les solubilisant ainsi dans les solvants organiques en les isolant de la phase organique mais aussi en les rendant en même temps accessibles. Le gabarit de la cavité peut être façonné pour permettre la séquestration spécifique et sélective de certains cations.
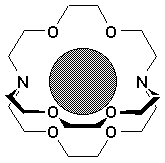
De plus, ce concept a été étendu en trois dimension avec la synthèse des cryptands (kryptos, grec, caché). La figure suivante montre un tel complexe (cryptate). Il sagit du cryptand 222, dont le nom systématique est: 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane.
Le système présenté se lie sélectivement à lion potassium ave une constante de liaison K valant 1010. Lordre de sélectivité est K+>Rb+>Na+>Cs+>Li+. La constante de liaison vis-à-vis du lithium est environ égale à 100. Ainsi, la valkeur de K relative à cette série de métaux alcalins sétend au total sur 8 ordres de grandeur!
5.3.11. Thiols et thioéthers (Vollhardt 9.10 et Arnaud 15.23 à 15.25)
Les thiols, (theion, grec, soufre natif) encore appelés mercaptans, et les thioéthers sont respectivement analogues des alcools et des éthers. Ici, un atome de soufre remplace latome doxygène.
RSH (thiol) RSR (thioéther)
On nomme les thiols en ajoutant le suffixe thiol après le nom, ou en utilisant le préfixe sulfanyl ou thio lorsque ce substituant nest pas le plus important:
| méthanethiol | CH3SH |
| éthane-1,2-dithiol | HSCH2CH2SH |
| 2-thioéthan-1-ol | HSCH2CH2OH |
Dans le cas des thioéthers, on utilise le terme sulfure:
CH3-S-CH2CH3 sulfure déthyle et de méthyle
Les thiols sont surtout reconnus pour leur odeur très nauséabonde. Ils peuvent être détectés à des teneurs aussi faibles que 0,02 ppb. Parmi les plus notoires, citons le H2S et les composés naturels qui comme lallicine (#36, Vollhardt p.323) donne lodeur alliacée de lail fraîchement coupé [Note]:
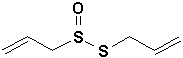
Le soufre étant moins électronégatif que loxygène, la liaison SH est moins polarisée que la liaison OH. Il en résulte des liaisons hydrogène nettement plus faibles que celles des alcools (CH3OH, Eb=64,7°C; CH3SH, Eb=6°C) (Tableau 9.2, Vollhardt p.305).
Les thiols sont toutefois plus acides que les alcools (pKa entre 9 et 12). Ici intervient surtout la grosseur de latome de soufre et son influence sur la longueur de liaison SH. De plus, il est plus apte à supporter les charges partielles positives et négatives. De plus, ils sont aussi de meilleures bases de Lewis.
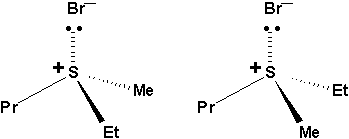
Par exemple, la molécule (CH3)3S+Cl est stable, alors que (CH3)3O+Cl na jamais été isolée. Ainsi, parce que latome de soufre est gros, la molécule suivante, le bromure de (R,S) éthylméthylpropylsulfonium présentera des isomères optiques:
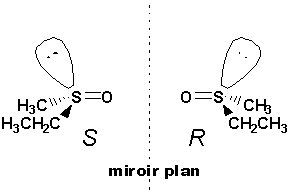
Ce phénomène est aussi rencontré chez les sulfoxydes:
Les thiols donnent donc plus facilement que les alcools des dérivés métalliques, les thiolates.
En ce qui concerne les réactions doxydation, elles sont très différentes de celles des alcools:
Un oxydant doux va conduire à la formation dun disulfure (principe de la coiffure permanente), parce que, contrairement à la liaison O-O (35 kcalmol-1), la liaison S-S (65 kcalmol-1) est très stable (p.307 en bas dans Vollhardt):
2 RSH + I2 ® RSSR + 2 HI
Lacide nitrique transforme les thiols en acides sulfoniques:
RSH ![]() RSO3H
RSO3H
Il est à noter que le groupe thiol et les ponts disulfures possèdent une grande importance dans divers processus biologiques comme la synthèse des protéines et des acides gras.
Les thioéthers peuvent être oxydés facilement en sulfoxydes et en sulfones:
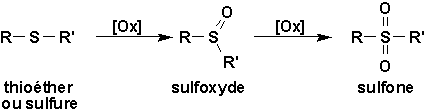
Finalement, il est à noter que de nombreux médicaments salutaires contiennent du soufre dans leur squelette moléculaire. Deux exemples sont présentés au milieu de la p.311 (Vollhardt). Citons entra autres les classes dagents bactériostatiques puisants connus sous les noms de sulfonamides ou sulfamides.
| Encarts du chapitre 9 Lire la section 9.11 On saute le reste du chapitre 9 Notions importantes 1 et 7 à la p.315. Le problème 13 des pages 315 à 323 peut être fait |
5.4. Amines (Vollhardt 21 et Allinger 4.14 à 4.16)
Les amines sont des composés dans lesquels un atome dazote est directement lié à un ou plusieurs atomes de carbone. Elles peuvent être considérées comme des dérivés de lammoniac, NH3 résultant de la substitution progressive des trois atomes dhydrogène par des groupes hydrocarbonés. Il existe donc 3 classes damines (primaire, secondaire et tertiaire):
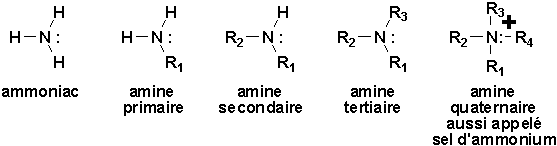
R1, R2 et R3 peuvent être identiques ou différents. Il est à remarquer ici que la convention primaire, secondaire et tertiaire des amines est différente de celle des alcools. Nous nous référons ici au degré de substitution de lazote et non pas à celui du carbone. par exemple, CH3CH(NH2)CH3 est une amine primaire de forme RNH2, alors que CH3CH(OH)CH3 est un alcool secondaire. Le véritable analogue structural oxygéné dune amine secondaire serait un éther.
À bien des égards, la chimie des amines est analogue à celle des alcools et des éthers. Ainsi, par exemple, toutes les amines sont basiques, encore que les amines primaires et secondaires peuvent aussi jouer le rôle dacides. Toutefois, parce que lélectronégativité de lazote est moindre que celle de loxygène, nous observerons certaines différences. De fait, les amines forment des liaisons hydrogène plus faible que les alcools.
5.4.1. Nomenclature des amines (Favre 5.4)
La nomenclature des amines est compliquée.
La façon la plus simple est de nommer les amines comme des alcanamines.| méthylamine | CH3NH2 |
| diéthylamine | (CH3CH2)2NH |
| tributylamine | (CH3CH2CH2CH2)3N |
| éthyl(méthyl)propylamine | (CH3CH2CH2)N(CH3)(CH2CH3) |
LUICPA recommande dutiliser le suffixe amine tout comme les suffixes ol et one. De plus, lorsque lamine nest pas le substituant principal, on utilisera le préfixe amino. La question de cette méthode est quil faut déterminer la chaîne principale. De plus, pour passer dune amine primaire à une amine secondaire ou tertiaire, il faut substituer latome dazote. Cette méthode est particulièrement utile pour les polyamines.
| éthanamine | CH3CH2NH2 |
| N-méthyléthanamine | CH3CH2NHCH3 |
| N,N-diméthyléthanamine | CH3CH2N(CH3)2 |
| N,N,N-triméthylbutane-1,4-diamine | (CH3)NH(CH2)4N(CH3)2 |
| 4-(2-aminoéthyl)heptane-1,7-diamine | Favre p.46 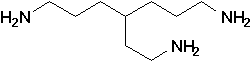 |
5.4.2. Caractères Physiques (Vollhardt 21.2 et Arnaud 17-1)
Les amines les plus légères, CH3NH2, (CH3)2NH, (CH3)3N, CH3CH2NH2, sont gazeuses à 20°C. Les autres sont des liquides ou des solides selon leur poids moléculaire.
Les liaisons NH donnent lieu à des liaisons hydrogène, mais à cause de lélectronégativité plus faible de lazote, ces liaisons hydrogène sont beaucoup plus faibles. Ainsi les points débullition des amines sont plus élevés que les hydrocarbures, mais moins élevés que ceux des alcools correspondants.
Les premiers termes conservent certaines des propriétés physiques analogues à celles de lammoniac, notamment leur grande solubilité dans leau et leur "odeur ammoniacale" caractéristique. De fait, les amines de poids moléculaire un peu plus élevées ont une forte odeur de poisson. Les amines benzéniques ArNH2 sont des liquides visqueux, ou des solides, insolubles dans leau et à lodeur désagréable.
Rappelons ici que la structure moléculaire des alcanamines est tétraédrique (Figure 21.1, Vollhardt p.826). Ceci implique que larrangement de substituants différents sur une amine devrait rendre celle-ci chirale et ainsi posséder une activité optique. Une telle conformation est montrée à la Figure au bas de la p. 826 dans Vollhardt. Toutefois, cette conformation nest pas stable, parce que lénergie dactivation nest que denviron 5 à 7 kcalmol-1, et lamine sinverse rapidement en passant par une hybridation sp2 (Figure 21.2 Vollhardt p.827). Ainsi, les amines trisubstituées énantiométriquement pures se racémisent rapidement à température ordinaire.
5.4.3. Réactivité (Arnaud 17-2)
Les liaisons CN et NH sont polarisées à cause de lélectronégativité de lazote. Dautre part, lazote possède un doublet libre. La situation est donc comparable à celle des alcools, mais étant donné que lazote est moins électronégatif que loxygène, la réactivité des amines sera donc différente de celle des alcools.
La disponibilité du doublet libre étant plus grande ici, les amines seront plus basiques que les alcools. La réactivité des amines se "concentre" sur lazote et son doublet libre, qui est pratiquement le seul site réactionnel.
Enfin, il existe de très grandes différences de réactivité entre les trois principales classes damines. La présence ou labsence dhydrogène sur lazote est fréquemment à lorigine de ces différences.
5.4.3.1. Propriétés acido-basiques (Vollhardt 21.4 et Arnaud 17.4 à 17.7)
Le comportement des amines primaires est double à cause de laccessibilité au doublet libre et de la labilité de lhydrogène.
Par exemple, en présence dun acide, une amine se comporte comme une base
RNH2 + AH ® RNH3+ + A
Alors quelle se comporte comme un acide en présence dune base
RNH2 + B ® RNH + BH
Le caractère acido-basique dominant des amines est nettement celui dune base:
R3N + HOH ® R3NH+ + OH
La constance déquilibre
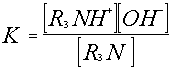
qui est la définition de la constante dacidité, Ka, du couple R3NH+/R3N vaut environ 10-10 (pKa = 10). Cela signifie que R3NH+ est un acide faible, et que R3N, sa base conjuguée, est une base moyennement forte. Les solutions des amines dans leau sont donc basiques (pH>7)
En règle générale, en phase gazeuse, la basicité des amines est gouvernée par les effets inductifs-répulsifs des substituants R, qui stabilisent lacide conjugué plus que la base en "comblant" partiellement le déficit électronique sur lazote:
NH3 < RNH2 < R2NH < R3N (< signifie "moins basique que")
En solution dans leau
, les amines secondaires sont les plus basiques, et les amines primaires et tertiaires ont sensiblement la même basicité. Les facteurs dencombrement stériques et laccessibilité du doublet par les molécules deau doit être pris en compte pour expliquer ce phénomène.5.4.3.2. Les sels dammonium quaternaires (Arnaud 17.9)
Les ions dammonium quaternaires sont obtenus en même temps quun anion, généralement un halogénure, avec lequel ils forment un sel dammonium quaternaire.
(CH3)3N + CH3Cl ® (CH3)4N+ Cl
Ces hydroxydes sont des bases aussi fortes que la soude ou la potasse, mais sont sensibles à laction de la chaleur.
Les sels dammonium tétrasubstitués énantiométriquement purs sont chiraux parce quil ny a plus de doublet libre présent pour permettre dinverser la molécule.
5.4.4. État naturel (Arnaud 17-3)
On retrouve la fonction amine dans les composés naturels au sein de structures complexes, et souvent associées à dautres fonctions. Citons notamment les alcaloïdes (atropine, caféine, cocaïne, morphine, nicotine, quinine). Elles interviennent aussi dans de nombreux mécanismes fondamentaux de la vie animale: acides aminés (constitutifs des protéines), bases azotés des acides nucléiques, neurotransmetteurs, régulateurs de la pression sanguine (adrénaline), etc.
5.4.5. Termes importants et utilisations (Vollhardt 21.12, encart 21.1 et Arnaud 17-5)
Sur le plan industriel, lamine simple, monofonctionnelle, la plus importante est laniline. Elle est notamment utilisée dans la fabrication des colorants, ainsi que dans celle des polyuréthanes.
Une diamine, lhexaméthylènediamine, H2N(CH2)6NH2, sert à la synthèse du Nylon. Dautres acides aminés sont à la base dautres textiles artificiels comme le Rilsan et le Perlon.
Les sels dammonium quaternaires sont utilisés comme agents tensioactifs et détergents, de même que comme bactéricides. Ils sont aussi utilisés en synthèse comme catalyseurs de transfert de phase.
Dans le domaine de la santé, on retrouve les éphédrines, amphétamines, théophyllines, décongestionnants, antidépresseurs et barbituriques (Encart 21.1). Aussi, certains alcaloïdes sont reconnus pour leur pouvoir hallucinogène: alcaloïdes de lopium, mescaline, acide lysergique (L.S.D.).
Finalement, on ne peut passer sous silence toute lindustrie des colorants qui doit énormément à la chimie de la famille des amines.
| Encarts du chapitre 21 On saute le reste du chapitre 21 Notions importantes 1 à 4 et 8 dans Vollhardt à la page 853 Les problèmes 1,2,7 des pages 853 à 860 peuvent être faits |
5.5. Organophosphorés (Allinger 4.14)
Tout comme lazote, le phosphore appartient au groupe 15 et possède 5 électrons de valence. Par contre, il nest pas limité à 5 liaisons covalentes. En effet, parce quil peut accommoder jusquà 10 électrons dans ses orbitales 3d il peut avoir, 3, 4, 5 ou 6 liaisons covalentes.
De plus, à cause de la taille de latome de phosphore, linversion des phosphanes est beaucoup plus lente que celle des amines. Il en résulte que les phosphanes asymétriques et les sels de phosphonium peuvent être séparés en leur énantiomères respectifs.
Du point de vue de la nomenclature, les organophosphorés se divisent en deux classes:
Les phosphanes et les l5-phosphanes
Les oxoacides
Les phosphanes et les l5-phosphanes sont les noms officiels de ce qui étaient auparavant les phosphines et les phosphoranes. Cette dénomination est très formelle (Favre, section 9.2.1).
| méthylphosphane | CH3PH2 |
| butyl(éthyl)méthylphosphane | Me-P(Bu)-Et |
| pentaéthoxy-l5-phosphane | P(OCH2CH3)5 |
Les oxoacides peuvent utiliser les nombres doxydation 3 et 5 du phosphore. La nomenclature est montrée dans Favre, section 9.2.2.
5.6. Organosilicés (Allinger 4.17)
Appartenant au groupe 14, tout comme le carbone, le silicium montrera une chimie similaire à son prédécesseur. Ainsi, nous observerons les types de composés suivants:
Les silanes, analogues des alcanes
| silane | SiH4 |
| disilane | H3Si--SiH3 |
| trisilane | H3Si-SiH2-SiH3 |
| tétraméthylsilane | (CH3)4Si |
| cyclohexasilane | 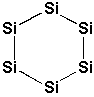 |
Les halogénures de silyle, analogues aux halogénures dalkyles
Les silanols, analogues des alcools
Les siloxanes, analogues des éthers
Les silazanes, analogues des amines
La nomenclature des silanes plus complexe est décrite dans Favre, section 9.3.
Le silicium possédant une structure tétraédrique comme le carbone, les silanes montreront aussi la possibilité de créer des molécules chirales.
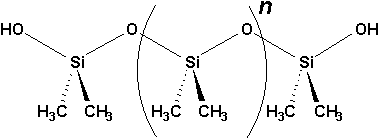
Compte tenu de ce que nous avons dit au début du cours concernant les énergies de liaisons, on ne sétonnera pas dapprendre que le silane et le disilane sont des composés qui senflamme spontanément à lair. Par contre, les liaisons SiO étant plus stables que celles du carbone, à cause des orbitales 3d vacantes du Si, une nouvelle catégorie de composés possédant une très grande stabilité thermique et servant dagents de lubrifiants verront le jour: les silicones.
5.7. Organoboriques (Allinger 4.18)
Jusquà maintenant, nous navons vu les structures des composés du carbone et du silicium quavec des éléments qui ne se retrouvaient quà droite du carbone dans la classification périodique.
Tous ces éléments comportent au moins un doublet délectron libre. ce sont des bases de Lewis.
Ainsi, les composés situés à gauche du carbone vont former des acides de Lewis à cause de leurs orbitales vacantes de basse énergie. On dit que ces molécules présentent une lacune électronique.
Parmi les composés du bore, nous retrouvons:
Les dérivés du borane, BH3
triméthylborane B(CH3)3
Les dérivés de lacide borique, B(OH)3
triméthoxyborane B(OCH3)3
Les dérivés de lacide boronique, HB(OH)2
Les dérivés de lacide borinique, H2BOH
Il est à noter que le bore a priorité sur le silicium:
silylméthylborane; H3Si-CH2-BH2
difluoro(triméthylsilyl)borane; (CH3)3Si-BF2
Cette nomenclature des boranes est décrite dans Favre, section 9.4.
Enfin, il est intéressant de noter que les hydrures de bore présentent la particularité la plus frappante de la chimie du bore. Lhydrure simple, BH3, est instable et se dimérise pour former le diborane, B2H6. Il existe de plus un certain nombre dhydrures supérieurs, par exemple B4H10 et B10H14. La structure du diborane est la suivante:
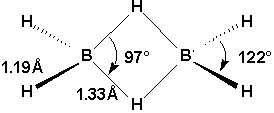
On remarquera quil est impossible de créer des structures de Lewis conventionnelles pour de tels composés, car les atomes dhydrogène seraient divalents!
Pour résoudre ce dilemme, il faut faire appel à la notion de liaison à plusieurs centres. Dans le diborane, chaque atome de bore est approximativement hybridé en sp3. Le recouvrement des orbitales créent ainsi une liaison à trois centres:
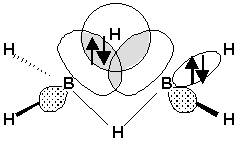
Ceci nest possible que lorsque lon a des atomes qui possèdent plus dorbitales de valence libres quil y a délectrons disponibles pour former des liaisons normales à deux centres.
5.8. Organométalliques (Allinger 4.19, Arnaud 14)
Les organométalliques sont des composés dans lesquels un métal est lié directement à un atome de carbone.
RCº CNa
est un organométalliqueRCONa
nen est pas un (cest un alcoolate)De nombreux métaux peuvent donner des combinaisons organométalliques, Li, Na, K, Mg, Cd, Zn, Hg, Al, Pb. Les métaux légers monovalents (Li, Na) sont les plus réactifs et donc les moins stables. Par exemple, les dérivés sodés du type RNa sont trop réactifs pour pouvoir être isolés, alors que le tétraéthyle plomb, Pb(CH2CH3)4, est un composé suffisamment stable pour être utilisé comme additif dans les carburants pour améliorer leur indice doctane.
Les organomagnésiens mixtes (ou réactifs de Grignard [Note]) possèdent comme formule générale RMgX. Ici, X est surtout Br, car Cl nest pas assez réactif et il est malaisé à préparer. Les organomagnésiens mixtes, pas plus que les organocadmiens, nexistent absolument pas à létat naturel, ce sont des composés synthétiques et qui, à leur tour, ne servent quà la synthèse, dans des réactions délicates.
La nomenclature est décrite dans Favre, chapitre 10. On utilise le radical approprié placé devant le nom du métal, sauf pour antimoine, Sb, bismuth, Bi, germanium, Ge, étain, Sn, plomb, Pb ou nous utilisons le nom de lhydrure parental mononucléaire.
butyllithium; CH3CH2CH2CH2Li
tétraéthylplumbane; (CH3CH2)4Pb
Dans le cas des coordinats anioniques, on cite, dans lordre: lanion suivi des groupes alkyles et du nom du métal, sauf pour antimoine, Sb, bismuth, Bi, germanium, Ge, étain, Sn, plomb, Pb ou nous utilisons le nom de lhydrure parental mononucléaire.
chlorure de méthylmagnésium; CH3MgCl
chlorodiéthylstibane; (CH3CH2)2SbCl
5.9 Exercices de nomenclature
Faire les exercices de nomenclature qui sont présentés dans Favre
Autres exemples: (réponses après les structures)
a) 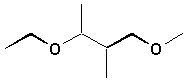 |
b) 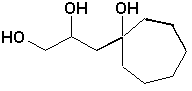 |
| c) |
d) 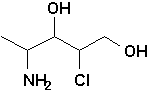 |
e) 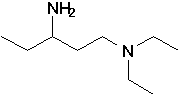 |
f) 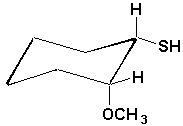 |
g) 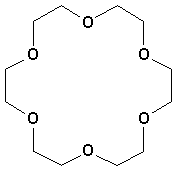 |
h) CH3CH2OCH(CH3)2 |
i) 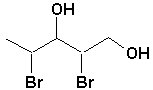 |
j) 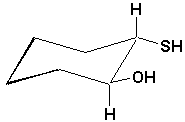 |
k) 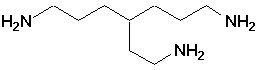 |
Réponses:
a) 3-éthoxy-1-méthoxy-2-méthylbutane
b) 3-(1-hydroxyxlcloheptyl)propane-1,2-diol
c) 1,2-bis(2-méthoxyéthoxy)éthane
d) 4-amino-2-chloropentane-1,3-diol
e) 1-(N,N)-diéthylpentane-1,3-diamine
(3-aminopentyl)diéthylamine
3-amino-N,N-diéthylpentyl-1-amine
f) cis ou (1S,2R)-2-méthoxycyclohexanethiol
oxyde de méthyle et de cis 2-thiocyclohexyle
g) éther-18-couronne-6
1,4,7,10,13,16-hexaoxacyclooctadécane
h) 2-éthoxypropane
2-méthyl-3-oxapentane
iso-propoxyéthane
oxyde déthyle et de méthyléthyle
oxyde d'éthyle et d'isopropyle
i) 2,4-dibromopentane-1,3-diol
j) trans ou (1S,2S)-2-thiocyclohexanol
k) 4-(2-aminoéthyl)heptane-1,7-diamine
| Retour au menu |
|
ABCSITE © copyright 2000 |