abcsite.free.fr
![]()
Chimie Organique
|
|
2. La Théorie structurale
| Dans ce chapitre | |
| 2.1. | Forces de Coulomb |
| 2.2. | Liaison ioniques et covalentes |
| 2.3. | Polarité et moment dipolaire |
| 2.4. | Structures de Lewis |
| 2.5. | Structures de résonance |
| 2.6. | Orbitales atomiques |
| 2.7. | Orbitales moléculaires |
| 2.8. | Orbitales hybrides |
| 2.9. | Composition, structure et formules des molécules organiques |
| 2.10. | Formule brute |
| 2.11. | Formules développées planes |
| 2.12. | Représentations simplifiées |
| 2.13. | Groupements fonctionnels |
| 2.14. | Composés à fonction multiples et mixtes |
| 2.15. | Groupes et radicaux |
| 2.16. | Premières notions de Stéréochimie |
2.1. Forces de Coulomb (Vollhardt 1.2)
Pourquoi deux atomes choisissent-ils détablir une liaison entre eux? Ils le font parce quil en résulte une condition favorable à cette nouvelle interaction.
Les bases des liaisons chimiques reposent sur le principe des forces de Coulomb, i.e. que des charges électriques éloignées de signe opposées sattirent mutuellement. Cette force dattraction entre les protons du noyau et les électrons qui les entoure est quantifiée par
F = cte ´
![]()
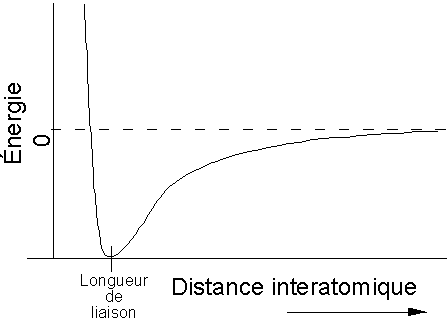
Cette attraction coulombienne entre les protons et les électrons force les atomes à se rapprocher pour former une liaison. Toutefois, à une certaine distance, elle est contrebalancée par la répulsion des nuages électroniques. Ainsi, il en résulte une position de minimum dénergie qui correspond à la longueur de la liaison entre les 2 atomes impliqués.
La position de ce minimum dépend de nombreux facteurs tels lélectronégativité, le rayon atomique, les orbitales impliquées et les charges en présence.
2.2. Liaison ioniques et covalentes (Vollhardt 1.3)
Je vous rappelle ici les définitions de ces deux types de liaisons:
La liaison ionique est réalisés lorsquun ou plusieurs électrons sont transférés dun atome à lautre, créant ainsi deux ions chargés: un cation chargé positivement et un anion chargé négativement. Cest ce que lon rencontre avec les sels inorganiques.
La liaison covalente résulte de partage mutuel des électrons entre les deux atomes.
Pourquoi y a-t-il ainsi deux sortes de liaisons? Revenons au tableau périodique. Certaines configurations électroniques sont particulièrement stables: il sagit des gaz rares. Cette classe déléments est caractérisée par une couche da valence entièrement remplis délectrons et une réactivité chimique nulle.
La règle de loctet veut que les atomes, considérés individuellement, réagissent dune manière telle quils peuvent remplir leur couche externe et ainsi atteindre une configuration électronique de type gaz rare. Cest ainsi que certains atomes vont perdre des électrons et dautres en gagner selon la position quils occupent dans le tableau périodique.
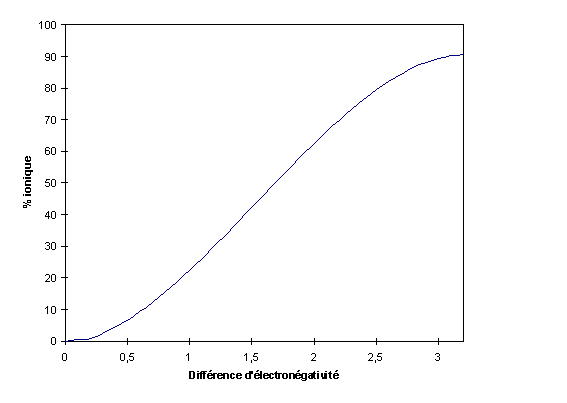
Toutefois, la vérité nest pas aussi simple que cela, car il nexiste pas de liaison 100 % ionique, et certaines liaisons covalentes possèdent un certain % ionique. En fait le % de liaison ionique dans nimporte quelle liaison dépend de la différence délectronégativité entre deux atomes, et ce de façon non linéaire. En fait, une première approximation serait:
% ionique = 33 ´ D électronégativité
La relation plus exacte est montrée ci-après:
% ionique = -4.935 D3 + 22.996 D2 + 5.748 D - 1.337
2.3. Polarité et moment dipolaire (Arnaud 8-2)
2.3.1. Polarité
Lorsquune liaison chimique unit deux atomes identiques, les électrons qui la constitue se retrouvent répartis au centre des deux atomes, à égale distance des deux et également partagés entre eux. (Le lecteur est ici prié de se remémorer les cours ce chimie de base).
Mais si ces deux atomes ne sont pas identiques, celui qui possède la plus forte électronégativité attirera vers lui les électrons plus fortement. Le nuage électronique ne sera plus symétrique et les charges électriques ne seront plus situées au centre des deux atomes. On dit dune telle liaison quelle est polarisée, car elle agit comme si elle possédait un pôle positif et un pôle négatif. On dit aussi quelle possède des charges électriques partielles positives et négatives.
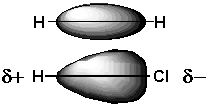
La liaison ne sera plus 100% covalente. mais possédera un certain caractère ionique. Toutefois, il faut se rappeler que même dans une liaison très polaire, par exemple Na+ et F- qui est la plus polaire imaginable, les charges partielles ne sont que ± 0,9.
2.3.2. Moment dipolaire
Létat de polarisation dune liaison est caractérisée par la valeur des charges d+ et d, mais aussi par le sens et la valeur de son moment électrique, ou moment dipolaire. Le moment dipolaire (m) est défini par la relation:
m = q ´ d
ou q est la charge absolue portés par chacun des atomes et d est la longueur de la liaison. On exprime les unités de moment dipolaire de Debye (D) [Note] selon:
m = 4.8 ´ q ´ d
lorsque q est exprimé en unités électroniques et d en nanomètres.
Le moment dipolaire est une quantité vectorielle, il possède un sens, une direction et un module. On le représente par une flèche parallèle à la liaison, orientée par convention du pôle + vers le pôle - (convention différente de celle des physiciens). une croix à lopposé de la pointe de la flèche rappelle cette convention.
![]()
Étant des quantités vectorielles, les moments dipolaires de toutes les liaisons dune molécule sadditionnent pour donner un moment dipolaire moléculaire global. Il est ainsi possible que ce moment dipolaire résultant ne coïncide avec aucune liaison. Il est aussi possible que la somme des moments dipolaires soit nulle. Ainsi une molécule comme le CCl4, qui possède 4 liaisons ayant des moments dipolaires important ne possède aucun moment dipolaire résultant à cause de sa symétrie. la molécule est alors dite non-polaire, et ce bien que ses liaisons soient polarisées.
Ce moment dipolaire est une caractéristique importante des molécules.
2.3.3. Effet inductif
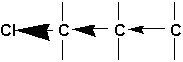
Leffet de la polarisation dune liaison peut se faire sentir à plusieurs atomes de distance. On dit dun tel effet quil est inductif. Ainsi, la présence dun atome fortement électronégatif comme le chlore se fait sentir sur le carbone qui le supporte, mais il influence les liaisons des deux atomes suivants:
Lélectronégativité de lhydrogène (2.20) étant voisine de celle du carbone (2.50), la liaison CH est très peu polarisés. Tous les autres substituants sont alors classés attractifs ou répulsifs par rapport à la liaison CH.
| Si certains dentre vous ont ici des problèmes, rapportez-vous aux sections 1.2 et 1.3 de Vollhardt. |
2.4. Structures de Lewis (Vollhardt 1.4)
On appelle structure de Lewis [Note] une représentation des atomes ou les liaisons sont caractérisées par des paires de points électroniques.
Les structures de Lewis obéissent à des règles simples.
Règle 1: Dessiner le squelette de la molécule.
Règle 2: Compter le nombre délectrons de valence disponibles. Une attention toute particulière doit être apportée aux structures chargées.
Règle 3 (règle de loctet): Représenter les liaisons covalentes entre tous les atomes en attribuant au plus grand nombre de ceux-ci un entourage électronique réalisant loctet, sauf pour lhydrogène qui ne nécessite quun doublet.
![]()
Ex:CCl4:1 C =4 e-C2H6:2
C =8 e-
4 Cl =28 e-6 H =6 e-
Total =32 e-Total =14 e-
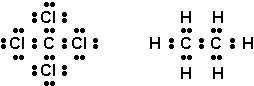
Il arrive aussi que des liaisons simples ne satisfassent pas la règle de loctet, la molécule N2 avec ses 10 e- de valence en est un exemple:
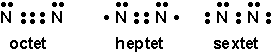
Règle 4: Attribuer les charges au sein de la molécule. Pour trouver les atomes qui porteront les charges, on ajoute tous les électrons de valence non impliqués dans des liaisons à la moitié des électrons de valence des liaisons contractées. Si le chiffre obtenu est égal au nombre délectrons de valence, la charge nette est nulle; si ces nombres ne sont pas les mêmes, latome est chargé.
Le calcul de la charge formelle est donc:
[ # e- valence ] - [ # e- non-liants ] - ½ [ # e- liens covalents ] = [ charge formelle ]
Par exemple, où se trouve la charge sur lion hydronium (hydron selon la nouvelle nomenclature, Favre 1992 p.127)?
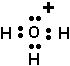
Ici, loxygène possède 3 liaisons simples et une paire délectrons non-liants. Il possède de plus 2 e- dans une couche sous-valentielle. Ainsi le calcul devient:
[ 6 e- ] - [ 2 e- non-liants ] - ½ [ 6 e- liens covalents ] = +1
Ce qui veut dire que cest loxygène qui porte la charge positive.
Létudiant est invité à tenter les exemples de la page 16 de
Vollhardt.
Cette façon décrire avec des points devient vite gênante, en particulier en présence de plus grandes molécules. Une façon plus simple est dindiquer les liaisons par de simples traits. Les paires délectrons libres sont, soit indiquées par des points, soit omises. Lusage de pareille notation a été proposée pour la première fois par un chimiste allemand Friedrich August Kekulé [Note] bien avant la découverte des électrons, de sorte que de telles structures sont souvent appelées structures de Kekulé.
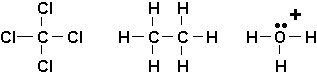
2.5. Structures de résonance (Vollhardt 1.5)
Nous allons maintenant rencontrer une série de molécules qui ne sont pas décrites par une structure de Lewis unique, mais par plusieurs. Comment est-ce possible?
Prenons lexemple de lion carbonate, CO32-. Daprès les règles énoncées pour les structures de Lewis, il est possible dimaginer une structure dans laquelle tous les atomes sont entourés par un octet délectrons. Toutefois, on saperçoit quil existe 3 choix possibles pour écrire la structure de la molécule:
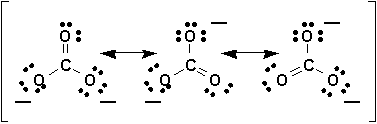
Mais il ny a pas de raisons particulières pour écrire un tel choix. Les trois atomes doxygène sont tous équivalents et indistincts les uns des autres. Les 3 structures sont équivalentes et on les appelle des structures de résonance. Les structures de résonance présentent la propriété caractéristique dinterconvertibilité, mais seulement par mouvement de paires délectrons, les noyaux devant rester inchangés au sein de la molécule.
Quelle est donc la véritable structure de lion carbonate? Si lon considère les trois structures ci-haut, et que lon imagine une moyenne des trois, on visualisera ce que lon appelle un hybride de résonance. Ainsi, la véritable forme de lion carbonate est la suivante:
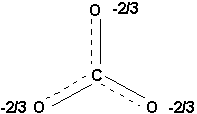
On peut montrer de nombreux exemples de structures de résonance comme celle-ci. Le benzène est dailleurs un cas particulier que nous traiterons en détail plus loin.
Mais quarrive-t-il lorsque la charge nest pas répartie sur des atomes identiques, comme par exemple dans lion énolate?
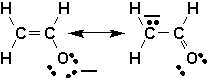
Étant donné que les deux structures sont différentes, lune dentre elles rendra mieux compte de la vérité que lautre, encore quaucune structure ne soit parfaitement correcte. Il existe des critères qui permettent de déterminer la stabilité relative de telles structures:
Critère 1: Les structures présentant un nombre maximum doctets sont préférentiels. Cest le cas de lion énolate, mais ce nest pas le cas pour le cation nitrosyle, NO+.
Critère 2: Les charges doivent être situées de préférence sur des atomes en concordance avec leur électronégativité. Dans le cas de lion énolate, cest ce qui arrive avec la première structure, loxygène étant plus électronégatif que le carbone. Toutefois, le critère 1 est plus important que le critère 2, comme cest le cas pour le cation nitrosyle, NO+ (Vollhardt p.19).
Critère 3: Les structures préférées sont celles où la séparation de charge est minimale. Cest une pure conséquence de la loi de Coulomb: il faut de lénergie pour séparer deux charges de signe opposées.
Critère 4: Les structures préférées peuvent être encouragées par la règle de loctet. Dans certains cas, créer une séparation de charge est le seul moyen de satisfaire au critère 1. Un exemple de cette situation est le monoxyde de carbone, CO (Vollhardt p.19).
2.6. Orbitales atomiques (Vollhardt 1.6)
Je vous rappelle rapidement ici que les électrons "orbitent" autour des noyaux selon la description de la mécanique quantique. La mécanique quantique a été développée dans les années 1920 par Werner Heisenberg (Munich), Erwin Schrödinger (Dublin) et Paul Dirac (Floride). Lorbite des électrons répondent à une fonction donde, et la représentation graphique de ces fonctions donde nous donne une image de la probabilité de retrouver lélectron dans une région donnée de lespace à un temps donné.
Nous nentrerons pas ici dans les détails de la mécanique quantique, les chimistes auront tout le loisir de la faire dans le cours de Chimie Théorique et Structure Moléculaire. Nous nutiliserons ici que le strict minimum applicable à la chimie organique.
Les orbitales atomiques représentent cette illustration graphique des fonctions donde qui répondent toutes à des solutions des équations de Schrödinger.
Elles sont caractérisées par les quatre nombres quantique:
nqui caractérise léloignement de le- par rapport au noyau
lqui caractérise la répartition dans lespace des orbitales [la dénomination des types dorbitales vient des raies spectrales: s=sharp, p=principal, d=diffuse, f=fundamental]
mqui caractérise la dégénérescence de lorbitale
squi caractérise le spin de le- dans lorbitale
La première solution est celle de lélectron tournant autour du proton dans latome dhydrogène; cest lorbitale 1s. Cette orbitale simple présente une symétrie sphérique, et ne possède pas de nud.

La solution suivante (et de plus haute énergie) est à nouveau unique et elle se traduit elle aussi par une forme sphérique: cest lorbitale 2s. Lorbitale 2s est plus grande que lorbitale 1s, due à lénergie plus élevée de lélectron. De plus, elle possède un nud au centre de la sphère.
Les solutions suivantes fournissent trois solutions équivalentes du point de vue énergétique: les orbitales 2px, 2py et 2pz.


Pour affecter les électrons à leur orbitales atomiques respectives, on doit utiliser léchelle de niveaux énergétiques suivant:
On ajoute les électrons un à un à la séquence orbitale telle quelle a été calculée pour latome dhydrogène; cest le Aufbauprinzip (Aufbau en allemand = construction) selon les trois règles suivantes:
Les orbitales de plus faible énergies sont remplies en premier.
Le principe dexclusion de Pauli stipule que deux électrons ne peuvent avoir les même nombres quantiques et occuper la même orbitale. Il sen suit quils auront donc des spins antiparallèles. On dit alors quils sont appariés.
La règle de Hund donne des instructions quant à la séquence selon laquelle les orbitales dégénérées, telles que les orbitales p, sont remplies: chacune dentre elles est dabord occupée par un électron, tous ceux-ci présentant le même spin. Sy ajoutent ensuite les autres électrons, avec un spin opposé à celui de la première série.
2.7. Orbitales moléculaires (Vollhardt 1.7)
Pour résumer une grande partie du cours de Chimie Théorique et Structure Moléculaire, il nous suffit de dire que les liaisons résultent du recouvrement en phase des orbitales atomiques. Il faut se rappeler que les orbitales sont des fonctions donde, et quelles peuvent interagir en se renforçant ou en sannulant mutuellement.
Il en résulte que laddition de 2 orbitales atomiques donne 2 orbitales moléculaires.
Si lon prend léquation de Schrödinger [Note]:
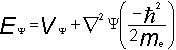
on peut effectuer une approximation des solutions pour les systèmes très simples comme latome dhydrogène. La première solution de létat fondamental de latome dhydrogène est celle de lélectron en orbite autour du proton et est décrite par lorbitale y1s centrée autour du noyau A. On la dénote 1sA. La fonction donde qui décrira la distribution globale de lélectron entre deux noyaux A et B est alors:
y = 1sA + 1sB
Il existe toutefois une seconde solution à léquation de Schrödinger:
y = 1sA - 1sB
Dans le premier cas, nous assisterons à la formation dune orbitale liante, et dans le second cas une orbitale anti-liante. On peut alors remplir ces orbitales selon le principe dexclusion de Pauli et la règle de Hund. Le tout est schématisé comme suit:
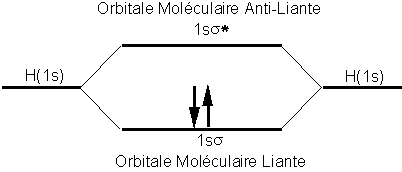
Lorsque lon fait le même exercice pour lhélium, on saperçoit que la molécule ne peut exister sous la forme diatomique car les orbitales anti-liantes annulent entièrement la contribution des orbitales liantes.
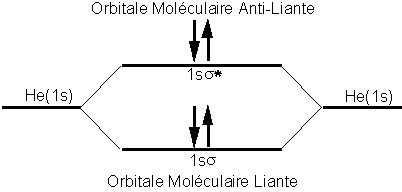
De plus, lintégrale de recouvrement, donnée par:
![]()
implique que certains recouvrements seront favorisés. Par exemple, le recouvrement linéaire s des orbitales p est plus forte que celle obtenue par un recouvrement latéral p de ces mêmes orbitales p. Ces orbitales présentant une faible intégrale de recouvrement posséderont par conséquent une énergie plus élevée (Figure 1.13, Vollhardt p.28).
On peut faire le même exercice que précédemment pour les orbitales supérieures et on obtient:
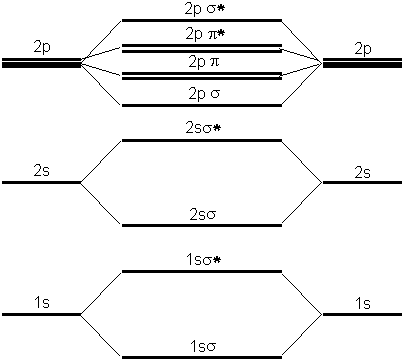
2.8. Orbitales hybrides (Vollhardt 1.8)
2.8. Orbitales hybrides (Vollhardt 1.8)
Considérons le carbone. Sa configuration électronique de base est (1s2) (2s2) (2p2) avec 2 électrons non appariés dans deux orbitales 2p.
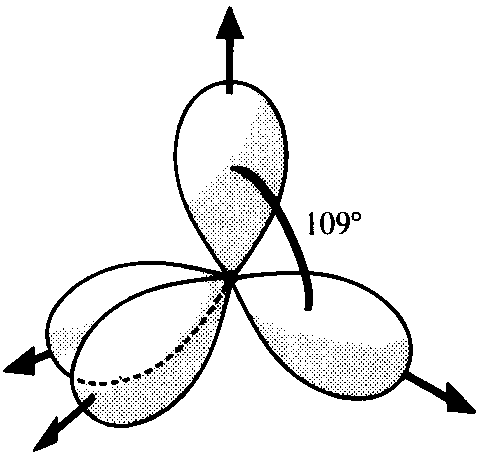
La promotion dun électron dune orbitale 2s vers 2p a pour conséquence la formation de 4 orbitales équivalentes, les orbitales sp3. Larrangement spatial de quatre charges dans lespace qui minimise la répulsion des charges sur une sphère imaginaire est celui du tétraèdre, ce qui explique la configuration en tétraèdre dune molécule comme le méthane, CH4, avec un angle de près de 109° entre chacune des liaisons.
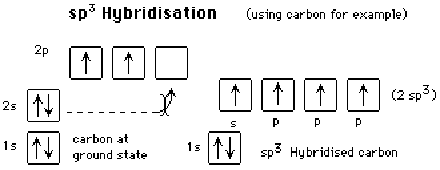
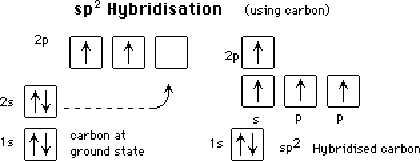
On peut utiliser lorbitale s et 2 orbitales p et former 3 orbitales hybrides sp2, qui seront alors séparées de 120°.

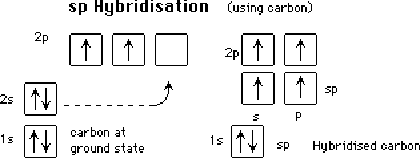
On peut utiliser lorbitale s et 1 orbitales p et former 2 orbitales hybrides sp, qui seront alors séparées de 180°.

2.8.1. Lorientation des liaisons autour des atomes de Carbone, Azote et Oxygène (VSEPR, Valence Shell Electron Pair Repulsing)
|
Carbone saturé |
4 liaisons simples |
tétraèdre |
109°28 |
|
Carbone éthylénique |
1 liaison double et 2 liaisons simples |
triangle équilatéral coplanaires |
120° |
|
Carbone acétylénique |
1 liaison triple et 1 liaison simple |
colinéaire |
180° |
|
Carbone allénique |
2 liaisons doubles |
colinéaire |
180° |
|
Azote saturé |
3 liaisons simples |
orientation pyramidale inscrite dans un tétraèdre dont la 4ième direction est occupée par le doublet libre |
109°28 |
|
Azote insaturé |
1 liaison double et 1 liaison simple |
orientation "en V" inscrite dans un triangle équilatéral dont la 3ième direction est occupée par le doublet libre |
120° |
|
Oxygène saturé |
2 liaisons simples |
orientation "en V" inscrite dans un tétraèdre dont les 3ième et 4ième direction sont occupées par les 2 doublets libres |
109°28 |
2.9. Composition, structure et formules des molécules organiques (Vollhardt 1.9, Allinger 2.2)
Du point de vue historique, il est important de se rappeler quen 1784, Antoine Lavoisier fut le premier à mesurer les résultats dune combustion. Il établit ainsi un rapport grossier C/H.
En 1831, Justus von Liebig [Note] réussit une combustion contrôlée. Cest la première analyse élémentaire connue. Les vapeurs organiques dune substance sont brûlées quantitativement sur du CuO chauffé au rouge. La réaction de la combustion de léthanol est la suivante:
C2H6O + 6 CuO + D ® 2 CO2 + 3 H2O + 6 Cu
6 Cu + 3 O2 + D ® 6 CuO
Le courant gazeux entraîne les gaz formés dans deux trappes successives. La première contient un agent desséchant comme le MgClO4, qui absorbe leau, puis la seconde trappe contient de la chaux sodée, CaO, qui transforme le CO2 en carbonate. En pesant les sels avant et après réaction, il est possible de calculer la quantité de carbone et dhydrogène que contenait la substance de départ.
Le seul élément quil est impossible de mesurer ainsi est loxygène, pour des raisons évidentes. Son abondance est obtenue par déduction.
À laide de cette analyse élémentaire il est possible détablir une formule brute du composé selon les relations suivantes:
H = 1.008 --- C = 12,01 --- O = 16,000
mH = mH2O ´
![]() % H =
% H = ![]() ´ 100
´ 100
mC = mCO2 ´
![]() % C =
% C = ![]() ´ 100
´ 100
% O = 100 - %H - %C (a priori)
Exemple: La combustion du méthoxyméthane donne les valeurs expérimentales suivantes de rapport massique:
%C = 52.24%H = 13.05
Des tests qualitatifs nous indiquent labsence de tout autre élément. La somme des masse négalant pas 100%, on en conclut que la substance contient 34.71% en poids doxygène. Ainsi, 100 g de substance contient 52.24 g de C, 13.05 g dH et 34.71 g dO. En tenant compte des masses atomiques il y a:
![]() =
4.36 atome-gramme de carbone
=
4.36 atome-gramme de carbone
![]() =
12.93 atome-gramme dhydrogène
=
12.93 atome-gramme dhydrogène
![]() =
2.16 atome-gramme doxygène
=
2.16 atome-gramme doxygène
Sans réfléchir beaucoup, on obtient la formule C4.36H12.93O2.16 (faire extrêmement attention aux arrondis, ils peuvent fausser les résultats pour de grosses molécules). Toutefois, on sait que le nombre datomes doit être entier. On doit donc diviser la structure par le plus petit nombre observé. On obtient alors C2.02H5.98O. À lerreur expérimentale près (moins de 0.3 % si lanalyse est correcte), la formule est donc C2H6O.
Ceci est donc la formule brute de notre composé, i.e. que lon a le rapport correct des éléments. La formule moléculaire est un multiple de celle-ci dans les mêmes proportions:
C2H6OC4H12O2C6H18O3
Ainsi, pour connaître cette formule réelle, on doit mesurer la masse moléculaire du composé.
Pour les gaz: mesures de P, V, T
Pour les liquides et solides: mesures cryoscopiques et ébullioscopiques dun solvant convenable.
Exemple: On veut effectuer lanalyse dune substance suspecte dont on recueille un échantillon de 1,234 g. À 190°C et sous 760 mm de Hg, léchantillon est vaporisé et occupe un volume de 410 cm3. La combustion complète de cet échantillon nous permet de recueillir 3,326 g de CO2 et 1,367 g dH2O (C=12.01, H=1.008, O=16.00). Déterminer:
A) sa composition centésimale
B) sa formule brute
C) sa formule moléculaire
D) Sachant par analyse RMN quil existe un groupement CH2OH et aucune liaison double sur cet molécule, et que de plus il y a 4 types de protons différents (intégration 1:4:4:2), quelle est la structure la plus probable de celle-ci et dites pourquoi? (Nous verrons la spectroscopie de résonance magnétique nucléaire plus loin. Toutefois, nous utiliserons un exemple ici pour démontrer le complément danalyse que cette technique nous permet)
A)mC = mCO2 ´ ![]() = 3.326 ´
= 3.326 ´ ![]() = 0.9076
= 0.9076
% C = ![]() ´ 100 =
´ 100 = ![]() ´ 100 = 73.55 %
´ 100 = 73.55 %
mH = mH2O ´
![]() = 1.367 ´
= 1.367 ´
![]() = 0.1530
= 0.1530
% H = ![]() ´ 100 =
´ 100 = ![]() ´ 100 = 12.40 %
´ 100 = 12.40 %
% O = 100 - %H - %C = 100 - 73.55 - 12.40 = 14.05 %
B)![]() = 6.124 atome-gramme de carbone
= 6.124 atome-gramme de carbone
![]() = 12.302 atome-gramme dhydrogène
= 12.302 atome-gramme dhydrogène
![]() = 0.878 atome-gramme doxygène
= 0.878 atome-gramme doxygène
La formule brute est donc C6.124H12.302O0.878
Divisant le tout par 0.878, on obtient: C6.975H14.011O qui devient: C7H14O
C)PV=nRT, 190°C=463K, 760 mm Hg=1 atm
conditions TPN 0°C, 1 atm, 1 mol, V=22.4 L : R
= ![]() =
= ![]()
![]()
= 1.08 ´ 10-2
Si 1.08 ´ 10-2 mol = 1.234 g, alors 1 mol = 114 g
C7H14O pèse 115.19, ce qui implique que la formule moléculaire est C7H14O
d)Si la molécule contient un groupement CH2OH, ceci implique quil reste C6H11 pour le reste de la molécule. Le spectre RMN ne montrant pas de protons du type alcène mais seulement des proton CH2. Il sagit donc fort probablement dun groupement cyclohexyle. Sa structure la plus probable est:
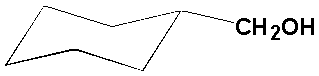
à cause des répulsions du cycle (voir démonstration du méthylcyclohexane plus loin), et aussi parce que le cycle à 6 est le cycle le plus stable.
La méthode développée par Liebig possède une précision de lordre de ±0.3%. Elle nécessite toutefois entre 500 mg et 1 g de produit. De telles quantité sont souvent difficiles à obtenir lorsque lon extrait ou synthétise des composés complexes. Une méthode de microanalyse basée sur la méthode de Liebig a été mise au point en 1911 par Fritz Pregl [Note] grâce à une amélioration considérable des instruments danalyse et plus particulièrement les balances. On peut ainsi opérer sur 3 à 4 mg de produit. Cette méthode a valu la prix Nobel à son inventeur en 1923.
De nos jours, on utilise de plus en plus le spectromètre de masse. Nous verrons cette méthode un peu plus en détail plus loin. Il faut dire quun spectromètre de masse coûte dans les 50 000 $ et plus...
Ainsi, nous venons de déterminer la formule brute et la formule moléculaire dun composé. Toutefois, ce nest pas suffisant. Il faudra utiliser des méthodes spectroscopiques comme la spectroscopie infrarouge, la résonance magnétique et la diffraction des rayons X pour nous permettre délucider les structures.
Comme mentionné précédemment, on entend par formule brute lénumération des différents éléments qui composent une molécule. Par exemple, la formule brute de lurée est CH4N2O.
Cela ne présente que peu dintérêt car elle ne nous donne pas dindice sur la façon dont les atomes sont liés entre eux. En fait, il existe 21 façon différentes non équivalentes dagencer ensemble ces 8 atomes.
Cela implique que nous devons faire appel à ce que nous appelons les formules développées.
2.11. Formules développées planes
Ces représentations sont dites planes car elles sont une représentation sur une feuille de papier de la structure réelle des molécules dans lespace. Elles représentent lordre dagencement de ces atomes, mais pas leur orientation réelle dans lespace.
Ainsi, si lon se souvient que le carbone possède une valence de 4, cela implique quil possédera 4 voisins. La structure réelle est celle dun tétraèdre et nous pouvons le représenter ainsi:
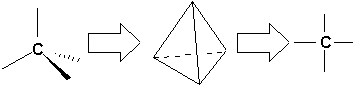
Il faut se rappeler que toutes les positions autour dun même atome de carbone sont équivalentes, cest lordre denchaînement qui est important. Ainsi les structures suivantes sont toutes équivalentes:
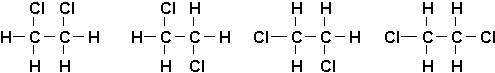
car chaque atome de carbone est relié à un autre atome de carbone, un atome de chlore et deux atomes dhydrogène. De plus, elles sont aussi différentes de:
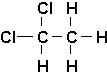
car ici un des atomes de carbone porte deux chlores, alors que lautre nen porte aucun.
La seule règle à suivre pour écrire correctement des formules planes est de donner à chaque atome le nombre de liaisons égales à sa valence, C=4, H=1, O=2, N=3, tel que:
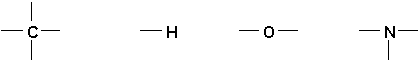
2.12. Représentations simplifiées
Ce sont des formules planes ou lon ne représente pas les atomes de carbone.
Par convention, il faut voir un C saturé au bout de chacun des segments et à leurs intersections. On ne représente que les autres hétéroatomes, et on sous-entend que les valences restantes des carbones sont comblées par des hydrogènes. De plus, dans le but de simplifier la représentation, les chaînes de carbone sont représentées par des lignes brisées:
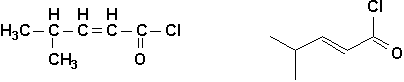
2.13. Groupements fonctionnels (Vollhardt 2.1)
On appelle groupement fonctionnel, tout groupe datomes qui, par leur arrangement particulier, confère des propriétés nouvelles aux molécules. Ce qui distingue les molécules entres elles cest justement la présence de ces groupes fonctionnels. Une molécule peut nen présenter quun, ou plusieurs. Théoriquement, il ny a pas de limites au nombre de groupements quune grosse molécule peut contenir.
Ainsi, nous avons par exemple les alcools (R-OH), les thiols (R-SH), les halogénures (R-X, ou X=F, Cl, Br, I), les amines (R-N<), les cétones (R-C=O), les acides carboxyliques (R-COOH).
Létudiant est prié de se référer au tableau 2.1 de Vollhardt aux pages 44 et 45. Toutefois, la nomenclature utilisée dans ce tableau nest pas conforme à la nouvelle norme. Ici comme ailleurs, La Nomenclature pour la Chimie Organique, publiée par lOrdre des Chimistes du Québec prévaut toujours.
2.14. Composés à fonction multiples et mixtes (Arnaud 20)
Il est fréquent dans les molécules organiques que lon rencontre plus dune fonction à la fois sur la même molécule. Il peut sagir de la même fonction (fonctions multiples), ou de fonctions différentes (fonctions mixte).
Que les fonctions gardent leur individualité ou soient modifiées dépend de la position relative des fonctions
Lorsque deux fonctions sont voisines, position a, il peut se produire dimportantes perturbations de leurs propriétés, (diène conjugué, énol).
Lorsquelles sont séparées par un atome de carbone, position b, leffet principal devient une activation des atomes compris entre celles-ci (par exemple la labilité des hydrogènes dun CH2 compris entre deux groupes carbonyles).
Lorsquelles sont séparées par une plus grande distance (4 ou 5 carbones au minimum) elles gardent leur individualité. Toutefois, il peut alors se produire un réaction intramoléculaire qui force la cyclisation de la molécule (la forme cyclique des sucres en est un exemple frappant).
Le terme de radical possède plusieurs significations.
Toutefois, nous emploierons le terme général de radical (R) pour désigner lhydrure parental qui est relié au groupement fonctionnel. Plus globalement, il signifie morceau de formule qui lon juge inutile de spécifier. Ainsi, les alcools possèdent la structure:
ROH
Il est inutile de décrire toutes les formes possible de R, car ce qui importe ici cest le groupement fonctionnel alcool, OH. On distingue toutefois deux types de radicaux:
| Les radicaux alkyles et cycloalkyles, désignés par la lettre R: | |
| CH3 | méthyle, Me |
| CH2CH3 | éthyle, Et |
| CH2CH2CH3 | propyle, Pr |
| CH2CH2CH2CH3 | butyle, Bu |
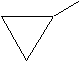 |
cyclopropyle |
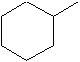 |
cyclohexyle |
| Les radicaux aryles, désignée par Ar | |
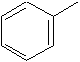 |
phénylePhe, f |
Le terme radical peut aussi signifier une molécule qui a perdu un électron et devient une espèce très réactive et instable. Toutefois, pour éviter toute confusion, il est plus correct dappeler ces dernières entités des radicaux libres.
2.16. Premières notions de Stéréochimie
Lorsque lon parle de stéréochimie, on parle de larrangement spatial des molécules. Toutefois, il est souvent difficile pour le néophyte de se représenter mentalement les molécules en trois dimension et encore plus ardu de lui demander de les faire tourner dans sa tête.
Pour ce faire, il faut faire appel à ce que lon appelle des projections. Nous allons introduire ici eux de ces projections.
2.16.1. Procédé du "coin volant" (flying wedge)
Ce type de projection utilise les trois liaisons suivantes:
| ------ | liaisons dans le plan du papier |
| - - - - | liaison orientée vers larrière du papie |
| coin noir | liaison orientée vers lavant du papier |
Ainsi, voici de quelles façon on peut représenter la molécule de méthane:
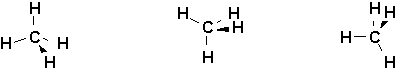
Il est a noter que lusage de modèles moléculaires aide souvent à bien se représenter ces projections.
2.16.2. Projection de Newman
La projection de Newman [Note] est souvent utilisé pour montrer la disposition relative des liaisons formées par deux atomes adjacents. Elle montre lorientation de ces liaisons telles quon les verraient si on regardait la molécule dans laxe de la liaison qui unit ces deux atomes.
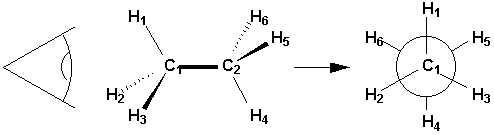
|
|
1.1.1. Les éléments constitutifs des molécules organiques (Arnaud 1.2)
Les éléments constitutifs des molécules organiques sont, par ordre de fréquence décroissant:
les quatre éléments, C, H, O, N
des non-métaux tels que Cl, Br, I, S, P, As, ...
des métaux tels que Na, Li, Mg, Zn, Fe, Co, Cu, Cd, Pb, Sn, ...
Comment compare-t-on labondance relative de ces éléments par rapport à ce que lon retrouve dans lunivers?
Doù ces éléments viennent-ils? Sans entrer dans les détails cosmologiques, on peut simplifier en disant que lors du Big Bang, lunivers formé contenait environ 93% H et 7% He. Les nuages dhydrogène se sont agrégés et ont formé la première génération détoiles. Les fournaises stellaires ont engendré les éléments plus lourds pour générer les éléments jusquau Fe. Ceci constitue une impasse thermodynamique et certaines étoiles, privées de leur carburant se sont alors effondrées et ont explosé (supernovæ) pour former les autres éléments au-dessus du fer.
|
Univers |
Terre |
Corps Humain |
|||
|
H |
93 |
O, oxydes et eau |
50 |
O, eau, protéines, phosphate |
65 |
|
He |
6.9 |
Si, silicates |
26 |
C |
18 |
|
O |
0.0005 |
Al |
7 |
H, eau |
10 |
|
C |
0.00008 |
Fe |
4 |
N |
3 |
|
N |
0.00015 |
Ca |
3 |
Ca |
2 |
|
Ne |
0.0002 |
Na |
2.5 |
P |
1 |
|
Autres |
0.1 |
K |
2.5 |
K |
0.35 |
|
|
|
Mg |
2 |
S |
0.25 |
|
|
|
H |
0.88 |
Na |
0.15 |
|
|
|
C |
0.087 |
Cl |
0.015 |
|
|
|
N |
0.030 |
Autres |
0.1 |
|
|
|
Autres |
2 |
|
|
Ainsi, à part loxygène qui est très abondant dans la croûte terrestre et le corps humain, les éléments C, H et N, qui constituent 31% du corps humain ne constituent que 1,78% des éléments présents dans lécorce terrestre et moins de 0,1% du total des éléments présents dans lunivers. Tout ceci implique que la chimie organique fait intervenir une très faible proportion des éléments connus.
Passons maintenant une très courte revue historique de la chimie organique.
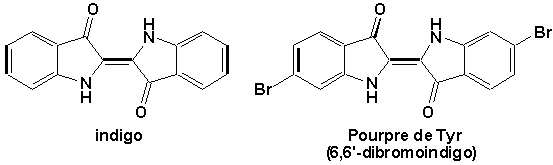
Dès lantiquité, les égyptiens, romains et phéniciens faisaient déjà de la chimie organique en réussissant à extraire des colorants naturels pour sen servir comme éléments de teintures pour leur vêtements. Citons notamment lindigo et lalizarine (provenant de la garance, une plante herbacée aux racines rougeâtre) et la pourpre de Tyr (provenant de coquillages) [Note]. L'industrie chimique organique origine des travaux de Perkin [Note] et de sa découverte du premier colorant synthétique: le Mauve de Perkins.
Des travaux importants ont été effectués dès 1923 par un savant russe du nom de A .I. Oparine [Note] ont portés sur latmosphère primitive de la terre. Il a ainsi proposé que celle-ci était composée dun mélange de H2O / CO2 / N2 / NH3 / CH4. Ceci a conduit Stanley L. Miller [Note] en 1950 a sa fameuse expérience où il a fait passer des décharges électriques dans un ballon contenant un tel mélange gazeux et ainsi former des acides aminés. Ce résultat fut extrêmement important car les protéines, des constituants majeurs des organismes vivants, sont faits dassemblages dacides aminés [Note].
1.1.2. Structure des molécules organiques (Arnaud, 0.4)
Les composés organiques et minéraux et leurs réactions chimiques respectives démontrent des particularités intéressantes. De plus, la position médiane du carbone dans la classification périodique des éléments et dans léchelle des électronégativités a pour conséquence que la chimie organique est essentiellement une chimie de composés covalents possédant des liaisons peu ou non polarisées.
|
Les Composés Organiques |
Les Composés Minéraux |
|
Sont formés de liaisons covalentes, ou à caractère covalent dominant |
Sont formés de liaisons ioniques, ou à caractère ioniques dominant |
|
Sont rarement solubles dans leau, et encore plus rarement des électrolytes |
Sont souvent des électrolytes, solubles dans leau |
|
Ont souvent des points de fusion et débullition bas, beaucoup sont des liquides à la température ordinaire |
Ont souvent des points de fusion et débullition élevés, beaucoup sont des solides cristallisés à la température ordinaire |
|
Ont le plus souvent une masse volumique voisine de lunité |
Ont des masses volumiques variables et souvent grandes (métaux) |
|
Sont facilement décomposé par la chaleur; peu résistants à une température supérieure à 500 °C |
Ont généralement une grande stabilité thermique (matériaux réfractaires) |
|
Sont presque tous des combustibles |
Sont rarement combustibles |
|
Les Réactions Organiques |
Les Réactions Minérales |
|
Sont souvent lentes, réversibles et incomplètes |
Sont souvent rapides et totales |
|
Ont le plus souvent des effets thermiques faibles (faible différence dénergie entre état initial et état final) |
Ont le plus souvent des effets thermiques forts (exothermiques ou endothermiques) |
1.1.3. Pourquoi le carbone? (Allinger 1)
Le carbone possède une configuration électronique 1s22s2p2 et donc 4 électrons de valence sur sa couche externe. ceci implique quil aura la possibilité de former jusquà 4 liens covalents pour compléter sa couche externe à 8 électrons (règle de loctet). La chimie particulière du carbone résulte de sa capacité à hybrider ses orbitales (on y reviendra sous peu).
Il est intéressant de noter quà cause de leur électronégativités voisines, les éléments N, H et O peuvent former des liaisons covalentes avec le carbone.
Accessoirement, on peut se demander si une chimie du même type que celle que lon observe avec le carbone est possible avec dautres éléments, notamment le silicium qui partage de nombreuses affinités électroniques avec le carbone: C [1s22s2p2] vs Si [1s22s2p63s2p2]. il faut ici regarder la force des diverses liaisons en jeux:
|
Lien |
kJmol-1 |
Lien |
kJmol-1 |
|
C-C |
610 |
C-H |
337 |
|
Si-Si |
327 |
C-O |
1077 |
|
Ge-Ge |
274 |
C-N |
770 |
|
Sn-Sn |
195 |
O-H |
428 |
|
Pb-Pb |
340 |
H-H |
436 |
|
O-O |
499 |
Si-O |
799 |
|
|
|
Si-H |
299 |
Ainsi, lénergie de liaisons de Si-O et C-C sont plus fortes que celles présentées par Si-Si. Ceci explique pourquoi les chaînes Si-Si sont détruites dans une atmosphère oxydante. Ainsi, les autres formes de vie que lon voit dans les films de science-fiction ne peuvent exister dans une atmosphère oxydante comme celle que nous possédons
| Retour au menu |
|
ABCSITE © copyright 2000 |